The target calculations ChemApp is able to perform are a very powerful tool to find one’s way around a phase diagram. The following program demonstrates, for the binary system Pb-Sn, how target calculations and standard equilibrium calculations can be used in the calculation of phase constitution. Figure 1 shows the binary phase diagram Pb-Sn, as it has been calculated using the two-dimensional phase mapping module of FactSage. The items labeled will be calculated in the course of this application example.
The type of information to be obtained should include the following:
- The determination of temperatures at which a phase (e.g. the liquid) becomes stable for an alloy of a given composition (formation phase target calculation, item 1 in Figure 1)
- Retrieving the necessary mole fractions and amounts which are associated with the ‘lever rule’ for an alloy at a given composition and temperature (item 2 in Figure 1).
- The determination of the temperature at which a phase (e.g. a solid phase from the liquid) starts to precipitate (precipitation target calculations, item 3 in Figure 1).
- Calculations similar to the ones above, but with composition instead of temperature as the target variable (item 4 in Figure 1).
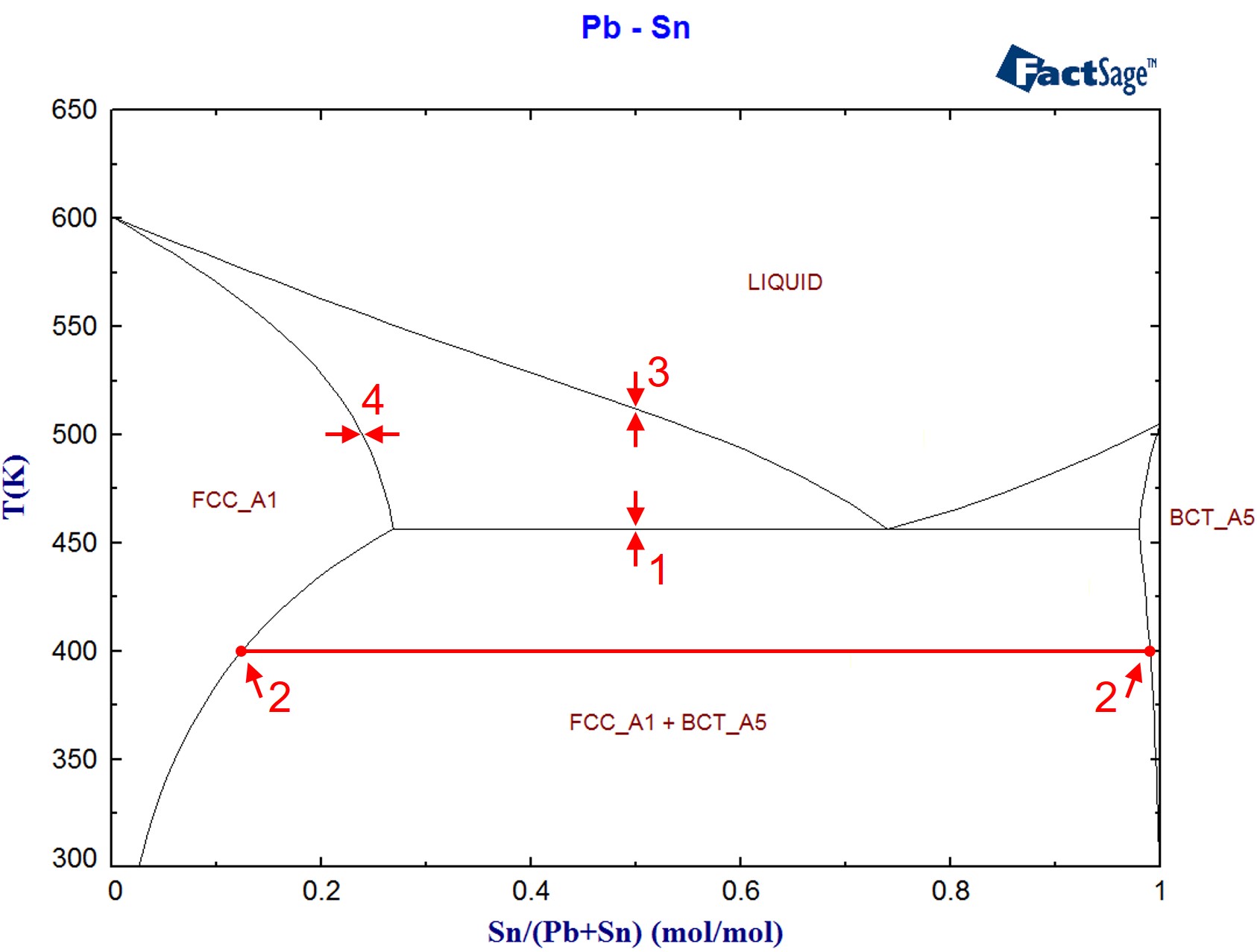
Figure 1: The Pb-Sn phase diagram (calculated using FactSage)
C Phase equilibria and phase target calculations C Note that this program contains target calculations, C which cannot be performed with the 'light' version of ChemApp PROGRAM CAF102 IMPLICIT NONE CHARACTER NAME*24, MODEL*6 DOUBLE PRECISION VALUE, VALS(2), FSNBCT, FSNFCC, * ABCT, AFCC, EPS INTEGER NOERR, ISLITE, NPHASE, NUMCON, I, NPCON, * IPB, ISN, I_LIQ, I_BCT, I_FCC C Initialise ChemApp CALL TQINI(NOERR) C Open the thermochemical data-file pbsn.dat (system Pb-Sn) C for reading OPEN(10, FILE='pbsn.dat', STATUS='OLD', IOSTAT=NOERR) IF (NOERR .NE. 0) THEN WRITE(*,*) 'ERROR: Cannot open data-file.' STOP ENDIF C Read data-file CALL TQRFIL(NOERR) C Close data-file CLOSE(10) C To find out about the phases contained in the data-file, print out the C names of all phases and the associated models CALL TQNOP(NPHASE, NOERR) DO I=1, NPHASE CALL TQGNP(I, NAME, NOERR) CALL TQMODL(I, MODEL, NOERR) WRITE(*,FMT='(I2,A)') I, ': ' // NAME // MODEL ENDDO
Output:
1: LIQUID RKMP
2: BCT_A5#1 RKMP
3: BCT_A5#2 RKMP
4: FCC_A1 RKMP
|
C As can be seen from the short table printed above, all phases present C in the data-file are mixture phases, since the model identifier C 'PURE' does not appear anywhere. Also, the phase BCT_A5 is entered C twice into the data-file. This is required because of the inherent C metastable miscibility gap in the system. C **************************************************************** C Formation phase target calculation C Item 1 in the Pb-Sn phase diagram C **************************************************************** C The first calculation will be the determination of the eutectic C temperature. To do this, we need to define an alloy composition which, C upon solidification, would undergo a eutectic transformation. An alloy C consisting of 50% Sn and 50% Pb is a good candidate. We will tell C ChemApp to use an alloy of this composition, and ask for the C temperature (which thus is the target variable) at which the liquid C phase first becomes stable (which thus is the target). Applying the C 'formation phase target liquid' thus means that ChemApp will determine C the point where the liquid phase is stable (activity is unity) and its C equilibrium amount is the one selected by the programmer (in our case C zero). C We will use 'global conditions', because we are not interested in C calculating extensive property (e.g. 'heat') balances. C We will use the system components to input our incoming amounts. Note C that it does not matter whether we input our incoming amounts as C system components, or through any of the mixture phase constituents or C stoichiometric compounds, since we are not interested in the extensive C property balances. C Find out the system component index numbers for Pb and Sn: CALL TQINSC('Pb ', IPB, NOERR) CALL TQINSC('Sn ', ISN, NOERR) C Using the index numbers just determined, we can enter our incoming C amounts: C Entering 0.5 mol Pb CALL TQSETC('IA ', 0, IPB, 0.5D0, NUMCON, NOERR) C Entering 0.5 mol Sn CALL TQSETC('IA ', 0, ISN, 0.5D0, NUMCON, NOERR) C Check if we are working with the 'light' version. C If we do, omit the following target calculation(s). CALL TQLITE(ISLITE, NOERR) IF (ISLITE .EQ. 1) THEN WRITE(*,FMT='(3(1X,A,/))') * '*** Target calculations have been omitted here,', * '*** since they are not possible with the ', * '*** ''light'' version of ChemApp.' ELSE C The 'LIQUID' phase is the target phase. From the first output we see C that it has the index number 1, which can also be determined by a call C of TQINP: CALL TQINP('LIQUID ', I_LIQ, NOERR) C Using TQSETC, we define the _target_: formation phase target 'LIQUID', C zero amount CALL TQSETC('A ', I_LIQ, 0, 0.D0, NUMCON, NOERR) C The _target variable_ is passed to TQCE/TQCEL: The target variable C used is the temperature, with 300 K as the first guess. This estimate C does not need to be very accurate. VALS(1) = 300.0 CALL TQCE('T ', 0, 0, VALS, NOERR) C Retrieve the calculated temperature CALL TQGETR('T ', 0, 0, VALUE, NOERR) WRITE(*,FMT='(1X,A,G12.5,A)') 'The eutectic temperature is ', * VALUE, ' K'
Output:
The eutectic temperature is 454.56 K
|
ENDIF C **************************************************************** C Phase equilibrium calculation (verification of the 'lever rule') C Item 2 in the Pb-Sn phase diagram C **************************************************************** C Now, for a temperature below the eutectic one, calculate the mole C fraction of Sn in both eutectic phases (FCC and BCT_A5#1) C The alloy composition is left unchanged (50% Sn, 50% Pb), but now the C temperature is set to 400 K CALL TQSETC('T ', 0, 0, 400.D0, NUMCON, NOERR) C Calculate the equilibrium CALL TQCE(' ', 0, 0, VALS, NOERR) C To check the lever rule for this case, and compare it to the graphical C representation in the Pb-Sn phase diagram, we need the mole fraction C of Sn in the BCT_A5#1 and FCC phases (FSNBCT and FSNFCC), the amounts C of BCC and FCC phase (ABCT and AFCC), plus the mole fraction of Sn in C the alloy, which is 0.5. C Get the equilibrium amount of phase BCT_A5#1 in mol CALL TQINP('BCT_A5#1 ', I_BCT, NOERR) CALL TQGETR('A ', I_BCT, 0, ABCT, NOERR) C Get the mole fraction of Sn in BCT_A5#1 CALL TQGETR('XP ', I_BCT, ISN, FSNBCT, NOERR) C Get the equilibrium amount of phase FCC in mol CALL TQINP('FCC ', I_FCC, NOERR) CALL TQGETR('A ', I_FCC, 0, AFCC, NOERR) C Get the mole fraction of Sn in FCC CALL TQGETR('XP ', I_FCC, ISN, FSNFCC, NOERR) C For the lever rule condition to be satisfied, the following equation C has to hold: (0.5 - FSNFCC)*AFCC - (FSNBCT - 0.5)*ABCT = EPS C with EPS being zero within the numerical precision. EPS = (0.5D0 - FSNFCC)*AFCC - (FSNBCT - 0.5D0) * ABCT WRITE(*,FMT='(1X,A,G12.5)') 'The value of EPS is ', EPS
Output:
The value of EPS is -0.48727E-15
|
C As you can see, the lever rule is sufficiently satisfied. C Check if we are working with the 'light' version. C If we do, omit the following target calculation(s). CALL TQLITE(ISLITE, NOERR) IF (ISLITE .EQ. 1) THEN WRITE(*,FMT='(3(1X,A,/))') * '*** Target calculations have been omitted here,', * '*** since they are not possible with the ', * '*** ''light'' version of ChemApp.' ELSE C **************************************************************** C Precipitation target calculation, with temperature as target C variable C Item 3 in the Pb-Sn phase diagram C **************************************************************** C The next calculation is again a target calculation. This time, a C 'precipitation phase target' is used to determine a point on the C liquidus curve, the alloy composition being the same as before. Using C a precipitation phase target tells ChemApp to search for the condition C under which a second phase becomes stable. C The subsequent call to TQSETC tells ChemApp to set the following C _target_: The liquid phase is stable, and a second phase (which does C not need to be specified) has unit activity. CALL TQSETC('A ', I_LIQ, 0, -1.D0, NUMCON, NOERR) C Use the temperature as _target variable_ and calculate the equilibrium VALS(1) = 1000.0 CALL TQCE('T ', 0, 0, VALS, NOERR) C Retrieve the calculated liquidus temperature CALL TQGETR('T ', 0, 0, VALUE, NOERR) WRITE(*,FMT='(1X,A,G12.5,A)') 'The liquidus temperature is ', * VALUE, ' K'
Output:
The liquidus temperature is 510.45 K
|
C **************************************************************** C Precipitation target calculation, with composition as target C variable C Item 4 in the Pb-Sn phase diagram C **************************************************************** C A target variable which is especially useful in T-x diagrams with C rather steep phase boundaries is the composition of an alloy. The C following example determines the point of the FCC phase boundary at C 500 K where LIQUID is formed (or precipitates). C First, for convenience, remove _all_ previously set conditions. This C is especially useful since we have to remove the conditions describing C the fixed composition of the alloy (set toward the beginning of the C program) anyway, as the composition is now the target variable and C thus supposed to vary. CALL TQREMC(-2, NOERR) C Set the temperature to 500 K CALL TQSETC('T ', 0, 0, 500.D0, NUMCON, NOERR) C Set the target: The FCC phase is stable, and a second phase has unit C activity. CALL TQSETC('A ', I_FCC, 0, -1.D0, NUMCON, NOERR) C If the composition is the target variable and the total input amount C should always be 1 mol, we need to vary two compositions C symmetrically. In the present case, we tell ChemApp to vary the input C amount of Sn/FCC/ between 0 and 1 to find the target, and to vary C Pb/FCC/ symmetrically between 1 and 0. C The upper and lower limits of the composition target variables are C passed via the array VALS to TQCE. First we pass the limits for C Sn/FCC/. VALS(1) = 0.0 VALS(2) = 1.0 C We need to tell ChemApp that we are not finished with our input, since C we still need to specify the symmetrical limits for Pb/FCC/ C later. This is done by calling TQCE with the option 'IA0', which tells C ChemApp not to perform an equilibrium calculation, but to expect more C incoming amounts. CALL TQCE('IA0 ', I_FCC, ISN, VALS, NOERR) C Enter the symmetrical limits for Pb/FCC/ VALS(1) = 1.0 - VALS(1) VALS(2) = 1.0 - VALS(2) C With the next call to TQCE, the information ChemApp needs to perform C a composition target is complete, so we call ChemApp with the option C 'IA', upon which the equilibrium is calculated. CALL TQCE('IA ', I_FCC, IPB, VALS, NOERR) C ChemApp has now varied the composition of the alloy until the desired C point of the phase boundary has been found. The desired value (the C mole fraction of Sn in the FCC phase) is retrieved. CALL TQGETR('XP ', I_FCC, ISN, FSNFCC, NOERR) WRITE(*,FMT='(1X,A,G12.5)') 'The mole fraction of Sn is ', FSNFCC
Output:
The mole fraction of Sn is 0.20698
|
ENDIF END
Continue with the code example “One-Dimensional Phase Mapping” or jump to the code example “Process Modelling Using Streams“.