ChemApp© - The Thermochemistry Library for your Software
© GTT-Technologies, Herzogenrath, Germany, 1996-2016
Programmer's Manual Edition 4.00 for ChemApp Versions 2.0.2 through 6.4.0

Authors: Gunnar Eriksson, Stephan Petersen
Address: Telephone: +49-2407-59533
Telefax: +49-2407-59661
E-mail: Web: Company: GTT-Technologies
Date: March 23, 2016
Edition: 4.00
Copyright:
Table of Contents
- 1 Introduction to the ChemApp Library
- 1.1 About this manual
- 1.2 Code examples
- 1.3 About ChemApp and ChemApp light
- 1.4 ChemApp is easy to program
- 1.5 Definition of terms
- 1.6 Introduction of concepts
- 1.7 Installation and use
- 1.8 Practical aspects
- 1.9 Language specific notes
- 1.10 ChemApp best practices
- 1.11 Thermodynamic data
- 1.12 Example thermochemical data-files
- 2 Initialising the Interface and Reading a Data-file
- 3 Handling System Data in ChemApp
- 3.1 Identifying phases, constituents, and components
- 3.2 TQINSC
- 3.3 TQGNSC
- 3.4 TQCNSC
- 3.5 TQNOSC
- 3.6 TQSTSC
- 3.7 TQCSC
- 3.8 TQINP
- 3.9 TQGNP
- 3.10 TQMODL
- 3.11 TQNOP
- 3.12 TQINPC
- 3.13 TQGNPC
- 3.14 TQPCIS
- 3.15 TQNOPC
- 3.16 TQSTPC
- 3.17 TQCHAR
- 3.18 TQINLC
- 3.19 TQGNLC
- 3.20 TQNOSL
- 3.21 TQNOLC
- 3.22 Changing the status of phases and constituents
- 3.23 TQGSP
- 3.24 TQCSP
- 3.25 TQGSPC
- 3.26 TQCSPC
- 4 Subroutines for Defining Equilibrium Calculations
- 5 Subroutines to Perform a Calculation and to Obtain Results
- 6 Data Manipulation Subroutines
- 7 List of Error Messages
- 8 Advanced topics
- 9 Worked examples
- 10 Thermodynamic Data - Sources and Quality Considerations
- 11 Thermochemical Data-files - Structure and Handling
- 12 FORTRAN example code (cademo1.f)
- 13 C example code (cademo1.c)
- 14 Example data-files and their contents
- 15 Revision history
- 16 Alphabetical list of ChemApp subroutines
- 17 Bibliography
1 Introduction to the ChemApp Library
ChemApp is a programmer's library consisting of a comprehensive set of subroutines, based on the thermodynamic phase equilibrium calculation module of ChemSage. It permits the calculation of complex, multicomponent, multiphase chemical equilibria and their associated extensive property balances. ChemApp is available as object code for a wide range of platforms and as a Dynamic Link Library (DLL) on Windows systems.
Potential applications for the use of ChemApp are almost limitless and can cover an extremely diverse range of applications. Two distinctly different groups of applications of ChemApp are possible:
- Using it for the development of application-specific programs; for example, for handling repetitive complex equilibrium calculations, for analysis, and for process control in well-defined technological areas.
- Linking it to third-party process simulation packages for modelling new or optimising existing processes; for example, commercial CFD programs such as Fluent®, CFX®, etc., general simulation programs, including Aspen Plus®, and also a company's own process simulation programs.
By embedding it in appropriate code, ChemApp can be employed to investigate time-dependent and kinetic effects using the concept of "local equilibria".
1.1 About this manual
Before starting to work with this manual, make sure the version of ChemApp you have matches the version of this documentation. This manual describes the use of ChemApp versions 2.0.2 through 5.0.2. If you are in doubt whether this is the right one in your case, please contact GTT-Technologies.
This documentation describes the regular, commercial version of ChemApp. A manual for our restricted, free version of ChemApp, called ChemApp light, is also available. This is referenced in About ChemApp and ChemApp light.
The next sections of the first chapter (ChemApp is easy to program to Language specific notes) deal mainly with the definition of terms, the introduction into concepts which are important to understand when using ChemApp, and some practical aspects in using the ChemApp library, including language-specific notes.
Section ChemApp best practices, is recommended reading for everbody who is looking at implementing anything but the simplest projects with ChemApp. It lists a number of very important programming issues to consider, which should be implemented as early in the stage of a ChemApp project as possible. If you encounter problems in your ChemApp programs and would like to contact GTT-Technologies' support, please make sure your code implements all of the "best pratices" outlined in this chapter before you send your code to GTT-Technologies' support.
The last parts of this chapter (Thermodynamic data and Example thermochemical data-files) describe which thermochemical data can be used with ChemApp and introduce the various example data-files that are supplied with ChemApp.
The actual ChemApp subroutines are described in Initialising the Interface and Reading a Data-file through Data Manipulation Subroutines, along with several dozen code examples in both FORTRAN and C, demonstrating their use.
A list of ChemApp error messages and their associated numbers is given in List of Error Messages.
Some advanced topics are discussed in the chapter Advanced topics. While the issues mentioned might not be vital for ChemApp programmers taking their first steps, anybody who is seriously interested in application programming using ChemApp should not omit reading it.
The chapter Worked examples presents a list of worked examples. Each example focuses on a different aspect of ChemApp and provides a good way to learn how ChemApp is used to solve a thermochemical problem.
Thermodynamic data and the handling of thermochemical data-files are treated in Thermodynamic Data - Sources and Quality Considerations and Thermochemical Data-files - Structure and Handling.
The next two sections contain the documented source as both FORTRAN (FORTRAN example code (cademo1.f)) and C (C example code (cademo1.c)) codes of a ChemApp example program which demonstrates the use of practically every ChemApp subroutine.
Finally, the chapter Example data-files and their contents lists the contents of all example data-files supplied with ChemApp, the section Revision history provides a revision history of this manual, and Alphabetical list of ChemApp subroutines contains a useful alphabetical reference of all ChemApp subroutines, to which the literature references (Bibliography) are added.
1.2 Code examples
This manual contains several dozen code examples to illustrate the use of the ChemApp subroutines and to support the method of learning-by-changing. Virtually all example codes are part of the distribution of the online manual, and you should feel encouraged to take these examples and start changing and editing them to take your own steps in programming with ChemApp. After receiving your distribution of ChemApp, it is reommended though to first run the program CADEMO1 and compare the results it produces on your computer with the master results which are normally included in the distribution. If you are using a FORTRAN or C distribution of ChemApp, you might also want to have a look at FORTRAN example code (cademo1.f) and C example code (cademo1.c) for that purpose.
The code examples have been designed to be both concise and easy to understand in terms of the ChemApp features involved. In terms of the application language used (FORTRAN and C), they require only basic knowledge. To keep these code examples short and easy to read, error handling has usually been neglected. In your own ChemApp programs, you should as early as possible adopt the policy to check the value of the error variable that every ChemApp subroutine returns.
The code examples used throughout this manual and the results they produce are not simply copied and pasted into the document. When the manual is produced, each code example is automatically compiled, linked to ChemApp, run, and checked for runtime errors reported by ChemApp. Only if no error occurred the code is mixed with the output produced during runtime, syntax-highlighted, and included into the manual. This guarantees that all example programs compile and run without problems with the ChemApp version that has been used to produce the manual. For the production of this edition of the manual Version 6.4.0 of ChemApp was used.
1.3 About ChemApp and ChemApp light
This is the documentation of the regular version of ChemApp. The use of your copy of ChemApp and its documentation is governed by the license agreement which you received with the object code, and which needs to be signed and returned to GTT-Technologies before you are permitted to start working with ChemApp.
A similar version of this documentation is also available for ChemApp light. This is a restricted version of ChemApp, which is distributed free of charge through GTT's Technical Thermochemistry Web Page.
One of the major reasons to produce ChemApp light was to provide potential users with an easy way to find out whether ChemApp can be linked to their own or third-party software. If you would like to introduce ChemApp to others, please refer them to ChemApp light, which can be downloaded from our web page.
If you have any questions regarding ChemApp, ChemApp light, this documentation, the code examples, or the example data-files, feel free to contact GTT-Technologies.
1.4 ChemApp is easy to program
Essentially, only three stages of programming are required to proceed from the initialisation of ChemApp to the collection of results; these are
- Initialisation of the interface and reading a thermodynamic data-file.
- Setting conditions for the calculation.
- Performing the calculation and collecting the results for further use.
In the simplest case, each stage can be accomplished by calling one or two subroutines. Naturally, each extra condition set, or result to be retrieved, increases the number of required subroutine calls.
1.5 Definition of terms
A thermodynamic system consists of a number of phases, where some may have a composition expressed as amounts of a number of phase constituents, and others can have an invariant composition. Phases are divided into three groups in ChemApp - the gaseous phase, condensed mixtures, and condensed stoichiometric phases.
Phases and phase constituents always have thermochemical properties (activities, chemical potentials, enthalpies, volumes, etc.). Phase constituents have compositions expressed as amounts (i.e. stoichiometric coefficients) of a number of components. A component is a system-wide entity. Sometimes this is stressed by calling it a system component. Usually components are elements, but it is also possible for them to be stoichiometric combinations of elements. For example, in an oxide system based on calcia and silica, CaO and SiO2 may be used, as well as Ca, Si, and O. Caution: In the more general case, it is necessary to ensure that the stoichiometric formulae are linearly independent.
Note: In a ChemApp data-file, the total number of components of a defined system includes phase-internal electrons; for example, when an aqueous phase is present. However, they are not counted when the maximum number of possible phases in the system is determined according to Gibbs' phase rule.
At chemical equilibrium, the absolute activities (chemical potentials) of the components are the same in the entire system. Internally in ChemApp, the actual number of system components is calculated as the rank of the stoichiometry matrix (see Thermochemical Data-files - Structure and Handling). This number may vary during the course of the Gibbs energy minimisation when only constituents with amounts greater than zero are considered. Should a complete reaction have taken place between two or more of the entered system components at equilibrium, it might not always be possible to calculate their absolute activities. For example, if in the system Mg-Si 2 mol of Mg and 1 mol of Si are entered, the stoichiometric condensed phase Mg2Si forms at low temperatures.
In order to calculate the thermochemical properties for the components, their respective reference state has to be selected. As this cannot be done in a general way, calculation of these properties is not included in ChemApp.
Components
ChemApp maintains a list of components. These are numbered sequentially from 1 up to the total number of components. This sequence of numbers will never change, even if only a subset is included in the input amounts of a calculation. Several subroutines are available for obtaining information on the components and how to manipulate them:
- TQNOSC:
- gets the total number of components
- TQINSC:
- gets the index number for a component
- TQGNSC:
- gets a component name
- TQSTSC:
- gets the stoichiometry and molecular mass of a component
- TQCSC:
- enables substitution of one set of components by another.
Phases
ChemApp maintains a list of phases. These are numbered sequentially from 1 up to the total number of phases. This sequential number does not change, even if phases are eliminated or set dormant.
- TQNOP:
- gets the total number of phases.
- TQINP:
- gets the index number for a phase
- TQGNP:
- gets a phase name
- TQMODL:
- gets the model name for a phase
Constituents
ChemApp maintains a list of the constituents of each phase. These are numbered sequentially from 1 up to the total number of constituents of each phase. This sequential number does not change, even if constituents are eliminated or set dormant. The number of constituents may be different in each phase. Stoichiometric condensed phases are considered to consist of one constituent only.
- TQNOPC:
- gets the total number of constituents of a phase
- TQINPC:
- gets the index number for a phase constituent
- TQGNPC:
- gets the name of a phase constituent
- TQSTPC:
- gets the stoichiometry of a constituent expressed in terms of the (presently defined) components, and its molecular mass
- TQPCIS:
- checks whether a phase constituent is permitted to be used as incoming species
Sublattices
ChemApp provides a number of subroutines to retrieve information on sublattices and their constituents.
- TQNOSL:
- gets the number of sublattices of a phase
- TQNOLC:
- gets the number of sublattice constituents of a sublattice
- TQINLC:
- gets the index number of a sublattice constituent
- TQGNLC:
- gets the name of a sublattice constituent
- TQGTLC:
- gets the calculated equilibrium sublattice site fraction
1.6 Introduction of concepts
In this section, several concepts are introduced which are important to the ChemApp application programmer. They include a description of the two ways initial conditions can be defined (global conditions and streams), and a short introduction to two calculational methods: target calculations and one-dimensional phase mapping.
1.6.1 Setting initial conditions
ChemApp offers considerable flexibility for defining initial conditions of a chemical equilibrium calculation. Two different methods are available that will cover most cases experienced in practice. Note, however, that you can only use one of these methods at a time; they cannot be used simultaneously.
Defining the global conditions of the system Using this method, you merely need to set single conditions for pressure and temperature, and enter incoming species to define the composition of the system. For example, if you wish to calculate the thermodynamic equilibrium for the system SiO2-CaO, using an appropriate thermochemical data-file that among others contains the elements Ca, Si, and O, you would only need to define the temperature and pressure of the system, and the total amounts of SiO2 and CaO present.
Conditions are set using the subroutine TQSETC and can be removed subsequently with the subroutine TQREMC. It is also possible to choose a number of other variables of state (see Table Possible state variables for setting conditions in the subroutines TQSETC and TQSTEC).
Defining streams A stream is a means for transferring non-reacted matter (see also the notes in Extensive properties of streams) to a reaction zone. It has constant temperature and pressure, and contains one or more phases of fixed composition. When using this method, you set the three variables - composition, temperature and pressure - for one or more input streams. For example, one stream entering a reaction zone may comprise O2(g) pre-heated to a temperature of 1500 K, and the other may consist of CO(g) at room temperature.
The streams method must be used for calculation of the extensive properties of reactions; for example, those involving the heat balance or the adiabatic temperature of a combustion process. It is also convenient to use it for reactor calculations, where you know what is entering the system and wish to calculate results at various stages during and at the end of the process. Streams are defined using the subroutine TQSTTP and can be removed again by using the subroutine TQSTRM.
The composition of a stream (see TQSTCA) must be entered using phases and phase constituents only. If one wants to enter the incoming amounts for a stream in terms of the system components (i.e. usually the elements), one has to perform an intermediate calculation. This is discussed in When phase constituents are not elements.
1.6.2 Target calculations
ChemApp is able to perform two types of target calculations:
- Extensive property target calculations
- Phase target calculations
An extension of the second type are one-dimensional phase-mapping calculations, see below.
Target calculations are those in which certain conditions (targets) are set, for instance the value of a heat balance or the presence of a phase at equilibrium. These conditions can only be satisfied when the value of some other variable, i.e. the target variable, is adjusted accordingly. When programming a target calculation with ChemApp, the target is set using TQSETC/TQSTEC, while the target variable is set when calling TQCE/TQCEL.
When performing an extensive property target calculation with ChemApp, one of the following five extensive properties of the system can be selected as target: Heat capacity, enthalpy, entropy, Gibbs energy, or volume.
A phase target calculation enables conditions to be determined when a selected phase is present at equilibrium (formation phase target), or when an unspecified phase is precipitated from a specified solution phase (precipitation phase target).
For examples on how target calculations are performed, see the code example for TQCE and the worked examples: Phase equilibria and phase target calculations and A simple process using streams.
Note that ChemApp light is not able to perform phase target calculations.
1.6.3 One-dimensional phase-mapping
One-dimensional phase-mapping is used to search for all phase transitions within a given interval of a search variable. The search variable can be either total pressure, temperature, or incoming amount.
For examples on how one-dimensional phase-mapping calculations are performed, see the code example for TQMAP and the worked example One-dimensional phase mapping.
Note that ChemApp light is not able to perform one-dimensional phase-mapping.
1.7 Installation and use
The regular version of ChemApp is supplied on disks or on CD, or is in special cases installed directly on your computer. In both cases, the code is in the form of readily usable libraries that can be linked to your application code with your own compiler. Please contact GTT-Technologies if you have any questions concerning the installation and use of ChemApp.
ChemApp is programmed in FORTRAN, as most large packages for fluid flow, process simulation, etc. were originally developed in this language. However, other languages can be used for the application program which links to ChemApp. In the present manual for instance, the use of C is demonstrated along with FORTRAN examples. For other supported languages like Delphi and Visual Basic, the distribution contains a port of the program CADEMO1, plus language specific application notes (see also below).
Starting with version 5.0.0, all Windows distributions of ChemApp require a hardware key (dongle). This is the same type of hardware key that is also required to run FactSage. In fact, ChemApp and FactSage can share the same physical dongle. If you are already a user of FactSage, you thus have the option of having your FactSage dongle(s) updated for use with ChemApp. Alternatively, or in case you are not already a user of FactSage, you also have the option of receiving separate dongles for use with ChemApp.
1.8 Practical aspects
The ChemApp library was designed to be strict and unambiguous in its use. Its purpose is to transfer information between the calling code and the internal calculation kernel, and to allow for the retrieval of results in a straightforward manner.
If ChemApp will be used with a language other than FORTRAN, please make sure you read the programming notes relating to that language. For your convenience these notes are included below (see Language specific notes). Please also read the notes on file that came with your ChemApp distribution, as they might contain more recent information.
Subroutine arguments
In the main part of this manual, all subroutines of the ChemApp library are described. Each subroutine contains a list of input and output variables or arrays as arguments. These are declared as either of type character, integer, double precision, or arrays thereof, and have to be passed or returned as one of these.
Character variables can be of any length in the application program, but are restricted to 24 characters in ChemApp. The exceptions are the subroutines TQERR, TQGTRH, TQGTID, TQGTNM, and TQGTPI, which all return character strings longer than 24 characters. Character options are case-insensitive. They can be entered preferably as arguments, but then may need to end with a blank space, depending on the compiler used. Both names and options can be uniquely abbreviated. When a character variable is output, it does not contain any leading blanks.
Entries for names of components, phases and constituents of a phase present in the thermodynamic data-file in use must be unambiguous. Names are case-sensitive and can consist of a string of letters up to 24 characters in length. For example, 'CO' and 'Co' can be two different phase constituents, and a component can be named 'My_favourite_component'.
Error messages
Whenever a syntax error in the call of a subroutine or a run-time error occurs, ChemApp returns an error number greater than zero via the last parameter of each ChemApp subroutine. It also writes an error message to the standard output unit. This destination unit, and thus also the file associated with it, can be changed using TQCIO, or turned off altogether. This is useful for application programs which make use of a graphical user interface (GUI), in which case the error messages can instead be selectively retrieved using TQERR.
It is strongly recommended to check the error number frequently and, if possible, to have the error message being displayed directly on screen whenever an error occurs (see for instance the FORTRAN and C versions of the demo program CADEMO1 in FORTRAN example code (cademo1.f) and C example code (cademo1.c)).
1.9 Language specific notes
The next sections contain language specific notes for ChemApp. They are also part of your ChemApp distribution. Please make sure you consult them, in case they contain more recent additions.
In the main part of the manual describing the ChemApp subroutines, reference is made not only to FORTRAN as a programming language, but also to C whenever possible. For readability purposes any specific C terms may have been omitted from the text, if the FORTRAN term used is unambiguous. For instance, when the FORTRAN term 'DOUBLE PRECISION' is used, it is understood that in C programming, 'double' is meant. The corresponding data types in all supported application languages are summarised in Table Data types used in ChemApp.
| FORTRAN | C/C++ | Delphi/Pascal | Visual Basic |
|---|---|---|---|
| INTEGER | long int | LongInt | Long |
| DOUBLE PRECISION | double | Double | Double |
| CHARACTER | char | String | String |
1.9.1 FORTRAN
- Avoid using names of variables, common blocks, subroutines, and functions starting with CS and TQ, as these might interfere with names used within ChemApp. If your compiler still produces warnings or errors that relate to variable and common block names starting with CS and TQ, your FORTRAN system libraries might make use of such names. In this case, please contact GTT-Technologies.
- All character variables for use with ChemApp which are supposed to hold names of system components, phases, phase constituents should be 24 characters in length.
Exceptions are the subroutine tqerr, as well as the subroutines tqgtrh, tqgtid, tqgtnm, tqgtpi, and tqwstr, which all pass or return strings longer than 24 characters.
1.9.2 C/C++
While ChemApp itself is written in FORTRAN77, the use of C/C++ for application programming is also possible on most platforms.
Most C/C++ compilers require an interface library for this purpose, in order to properly link to the ChemApp library. This interface consists of
- either a readily compiled library (e.g. libChemAppC.a), or a source code file (e.g. cacint.c), depending on your particular distribution, which you add to your projects,
- a header file (e.g. cacint.h).
Please refer to the C example supplied with this distribution for information on how to use these files. The header file needs to be included by your C/C++ files, and the library needs to specified during the link process.
Here is a list of points to be aware of when using ChemApp with C/C++:
- C/C++ is case-sensitive in contrast to FORTRAN. When calling ChemApp functions, please make sure you use lowercase function names (e.g. use tqini, not TQINI).
- There is one ChemApp function which takes an array of strings as input parameter: tqcsc (Change-System-Components). Due to the differences between the FORTRAN and C/C++ parameter passing and string handling, C/C++ code has to provide a slightly different string array at this point than FORTRAN code. Whereas in FORTRAN this array has to consist of as many strings as there are system components, in C/C++ it has to have one extra string element, which has to be empty. Please see the C program provided for an example usage of tqcsc.
- Please keep in mind that in C/C++, arrays start with element number 0, whereas in FORTRAN, they normally start with 1. This often needs to be taken into account when arrays are involved in loops which contain ChemApp routines to access a range of index numbers or names of system components, phases, and phase constituents.
- As opposed to FORTRAN77 character variables, which are fixed in length and padded with trailing blanks if necessary, strings in C/C++ are null-terminated. The C interface routines delete trailing blanks from strings returned by ChemApp routines and add a null character at the end, before passing them back to the calling C/C++ program. Similarly, they process a C/C++ string passed to ChemApp in order to constitute a proper FORTRAN character variable of the right size before passing it on to the ChemApp subroutines. In order to do this without flaws, please make sure you declare the C string variables and arrays in your C/C++ programs correctly, i.e. giving them the right length. The C example program provided demonstrates several ways how to do this safely.
All character variables for use with ChemApp, e.g. variables that hold names of system components, phases, phase constituents as well as for other purposes like identifiers for the status of a phase, should be 24 characters in length (this means 25 characters in C/C++, taking the trailing null character into account).
Exceptions are the subroutine tqerr, for which a variable TQERRMSG is declared in cacint.h, as well as the subroutines tqgtrh, tqgtid, tqgtnm, tqgtpi, and tqwstr, which all pass or return strings longer than 24 characters.
- On some platforms, a FORTRAN INTEGER variable is different in size from a C/C++ int variable. The same is possible for FORTRAN's DOUBLE PRECISION and C/C++'s float. On most platforms, INTEGER corresponds to long int and DOUBLE PRECISION corresponds to double. To avoid compiler error messages due to these differences, please use the type LI when declaring an integer variable for use with ChemApp in C/C++, and DB when declaring a float variable for use with ChemApp. These two types are defined in cacint.h specific to your platform. See the C program provided for examples.
- If you get warnings like "... makes pointer from integer without a cast" when compiling your C/C++ code, check whether you specified the address operator (&) properly when passing variables to ChemApp functions. All variables through which values are returned, and which are not considered arrays by C/C++, need the address operator.
- The ChemApp subroutines tqrfil (Read-File), tqrbin (Read-Binary-Data-File), and tqrcst (Read-Transparent-Data-File) expect the file from which the thermodynamic data should be read to be open. In FORTRAN, the standard statement OPEN is used to open a file using the correct unit number. In non-FORTRAN programs, there is no way to make sure a file opened with functions such as fopen gets associated with a certain unit number so that a FORTRAN subroutine can access it. To overcome this problem, special FORTRAN routines are provided which open and close a file using C/C++ under a specific FORTRAN unit number. Refer to the C program provided and the documentation to see how these functions (tqopna, tqopnb, tqopnt, tqopen and tqclos) are used.
Note that tqopen does not check whether the file to be opened already exists or not, since tqopen can be used both for opening existing files for reading, and new files for writing (tqcel, tqshow). Whether the file to be opened already exists should be checked prior to a call to tqopen. Also, the use of tqopna, tqopnb, and tqopnt instead of tqopen is strongly recommended to open thermochemical data-files for reading.
1.9.3 Delphi
While ChemApp itself is written in FORTRAN77, the use of Borland Delphi® for application programming is possible by using the ChemApp DLL for Borland Delphi.
Please note that the DLL versions of ChemApp are usually not interchangeable between the compilers. To utilise ChemApp with your Delphi projects, you need to make sure that you are using the ChemApp DLL for Borland Delphi, a ChemApp DLL for another compiler (e.g. Microsoft Visual C++®) will likely not work.
To make use of ChemApp from within your Delphi projects, you need to
include the file cadint.pas in your projects (this file contains the Delphi interface to ChemApp and is part of the distribution),
For ChemApp versions 5.0.0 and later: make sure that in the project options of your application, you use one of the following conditional defines to indicate which version of the ChemApp DLL your application should use.
If you would like to use the standard version of ChemApp, use the conditional CA_DEL_S. This will result in your application making use of ca_del_s.dll.
If you would like to use the extended version of ChemApp, use the conditional CA_DEL_E. This will result in your application making use of ca_del_e.dll.
If you would like to use the light version of ChemApp, use the conditional CA_DEL_L. This will result in your application making use of ca_del_l.dll. This is also the default, i.e. the light version is used when none of the other conditionals is defined.
For ChemApp versions less than 5.0.0: make sure that the ChemApp DLL for Delphi is available, and its name is assigned to the constant ChemAppDLL in the file cadint.pas (depending on the distribution you are using, ChemApp comes in a "standard", "extended", or "light" version, which are named differently to avoid ambiguity).
Please use the Delphi example program supplied with this distribution as a test case to determine whether you set up your projects correctly. Compare the results this program generates to the master results in the files cademo1.rst and result. If you have any problems reproducing these results, please contact GTT-Technologies.
Here is a list of points to be aware of when using ChemApp with Delphi:
- There is one ChemApp function which takes an array of strings as input parameter: tqcsc (Change-System-Components). Due to the differences between the FORTRAN and Delphi parameter passing and string handling, Delphi code has to provide a slightly different string array at this point than FORTRAN code. Whereas in FORTRAN this array has to consist of as many strings as there are system components, in Delphi this array has to have one extra string element, which has to be empty. Please see the Delphi program provided for an example usage of tqcsc.
- Every "name" used in conjuction with ChemApp, whether it is the name of a phase, a system component, a phase constituent, or a stream, can be up to 24 characters in length.
In contrast to other languages, Pascal is strict in terms of parameter type matching, which makes it difficult to use a type like string[24] for this purpose (as is done in the C/C++ interface to ChemApp).
Thus, all strings used in the Delphi interface to ChemApp (cadint.pas) are of type string, which means that all strings declared in an application program and used as parameters for ChemApp procedure calls should be of type string, or otherwise properly typecast/converted. It is important to realize that for names like system components, phases, phase constituents, etc., ChemApp itself does only make use of the first 24 characters of a string passed to it, and only returns a maximum of 24 characters to the calling code.
Exceptions are the subroutine tqerr, for which the type Terrmsg is declared in cadint.pas, as well as the subroutines tqgtrh, tqgtid, tqgtnm, tqgtpi, and tqwstr, which all pass or return strings longer than 24 characters.
- Every integer variable to be used for ChemApp procedure calls needs to be of type longint, every floating point variable of type double. To simplify the declaration of those variables, and to make the Delphi version of the interface to ChemApp consistent with that in other languages, several "type"s have been declared in cadint.pas, like LI (for longint) and DB (for double), which you are encouraged to use. Please see the type declaration section in cadint.pas for a list of these types.
- The ChemApp subroutines tqrfil (Read-File), tqrbin (Read-Binary-Data-File), and tqrcst (Read-Transparent-Data-File) expect the file from which the thermodynamic data should be read to be open. In FORTRAN, the standard statement OPEN is used to open the file using the correct unit number. In Delphi, there is no way to make sure a file opened with functions such as AssignFile gets associated with a certain unit number so that a FORTRAN subroutine can access it. To overcome this problem, special FORTRAN routines are provided which open and close a file using Delphi under a specific FORTRAN unit number. Refer to the Delphi example program provided to see how these functions (tqopen, tqopna, tqopnb, tqopnt, and tqclos) are used.
Note that tqopen does not check whether the file to be opened already exists or not, since tqopen can be used both for opening existing files for reading, and new files for writing (tqcel, tqshow). Whether the file to be opened already exists should be checked prior to a call to tqopen, tqopna, tqopnb, and tqopnt. For further details, see the manual section on tqopen.
1.9.4 Visual Basic
While ChemApp itself is written in FORTRAN77, the use of Microsoft Visual Basic® for application programming is possible by using the ChemApp DLL for Microsoft Visual Basic.
Please note that the DLL versions of ChemApp are usually not interchangeable between the compilers. To utilise ChemApp with your Visual Basic projects, you need to make sure that you are using the ChemApp DLL for Microsoft Visual Basic, a ChemApp DLL for another compiler (e.g. Microsoft Visual C++®) will likely not work.
To make use of ChemApp from within your Visual Basic projects, you need to
- include the module cavbint.pas in your projects (this file contains the Visual Basic interface to ChemApp and is part of the distribution),
- make sure that the version of the ChemApp DLL for Visual Basic you intend to use is available, and the matching conditional compilation directive (conChemAppStandard, conChemAppExtended, or conChemAppLight) is set to 1 in the file cavbint.pas (ChemApp is supplied in "standard", "extended", and "light" versions, which are named differently to avoid ambiguity).
Please use the Visual Basic example project cademo1 supplied with this distribution as a test case to determine whether you set up your projects correctly. Compare the results this program generates to the master results in the files cademo1.rst and result. If you have any problems reproducing these results, please contact GTT-Technologies.
Here is a list of points to be aware of when using ChemApp with Visual Basic:
- Which version of the ChemApp DLL ("Standard", "Extended", or "Light") is to be used when a program is compiled and run is determined using conditional compilation directives. The proper directive (conChemAppStandard, conChemAppExtended, or conChemAppLight) can either be set through cavbint.bas (as described above), or by conditional compilation directives specified in the Project Properties dialog box, or on the command line.
For more information on this topic, see the section "Using Conditional Compilation" in the "More About Programming" section of the "Visual Basic Programmer's Guide".
- There is one ChemApp function which takes an array of strings as input parameter: tqcsc (Change-System-Components). Due to the differences between the FORTRAN and Visual Basic parameter passing and string handling, Visual Basic code has to provide a slightly different string array at this point than FORTRAN code. Whereas in FORTRAN this array has to consist of as many strings as there are system components, in Visual Basic this array has to have one extra string element, which has to be empty. Please see the Visual Basic program provided for an example usage of tqcsc.
- Every "name" used in conjuction with ChemApp, whether it is the name of a phase, a system component, a phase constituent, or a stream, can be up to 24 characters in length.
FORTRAN77 only knows strings of fixed length. Using the same type of strings on the Visual Basic side of the interface is unpractical, since every string variable returned by ChemApp would be padded with blanks. Thus, all strings used in the Visual Basic interface to ChemApp are variable-length strings (Dim As String), with trailing blanks properly removed when returning from ChemApp. It is important to realize that ChemApp itself does only make use of the first 24 characters of a string passed to it, and only returns a maximum of 24 characters to the calling code.
Exceptions are the subroutine tqerr, for which a variable TQErrMsg is declared in cavbint.pas, as well as the subroutines tqgtrh, tqgtid, tqgtnm, tqgtpi, and tqwstr, which all pass or return strings longer than 24 characters.
- Every integer variable that is to be used in an invocation of a ChemApp subroutine has to be of type Long. Similarly, every real variable for use with ChemApp subroutines has to be of type Double.
- The ChemApp subroutines tqrfil (Read-File), tqrbin (Read-Binary-Data-File), and tqrcst (Read-Transparent-Data-File) expect the file from which the thermodynamic data should be read to be open. In FORTRAN, the standard statement OPEN is used to open the file using the correct unit number. In non-FORTRAN programs, there is no way to make sure a file opened with functions such as fopen gets associated with a certain unit number so that a FORTRAN subroutine can access it. To overcome this problem, special FORTRAN routines are provided which open and close a file using C/C++ under a specific FORTRAN unit number. Refer to the C program provided and the documentation to see how these functions (tqopna, tqopnb, tqopnt, tqopen and tqclos) are used.
Note that tqopen does not check whether the file to be opened already exists or not, since tqopen can be used both for opening existing files for reading, and new files for writing (tqcel, tqshow). Whether the file to be opened already exists should be checked prior to a call to tqopen. Also, the use of tqopna, tqopnb, and tqopnt instead of tqopen is strongly recommended to open thermochemical data-files for reading.
1.10 ChemApp best practices
Like any other tool, software or otherwise, ChemApp can be used in a good way, and in a better way. The following section intends to familarize you with guidelines to help you write better code with ChemApp, and in doing so should aid you in achieving your objectives wasting not only less time, but also less nerves.
If you encounter a problem in your work with ChemApp, and would like to contact GTT-Technologies' support in order to help you track down a problem in your code, or find out whether the problem might lie in ChemApp itself, this section is recommended reading. The points below are the result of several years of experience with many support issues, and we thus strongly recommend checking your code as to whether these best practises are implemented before you send it to us.
In fact, we have found that many apparent problems turn out to be "false alarms" on the user's side, and become obvious once they follow these best practices. Due to the time constraints and the workload of GTT-Technologies' support team, we might have to refrain from debugging users' code if these best practices are not implememted. (A typical example is that the error code which every ChemApp subroutine returns is only checked when calculating the equilibrium with TQCE, thus overlooking the fact that the actual reason why an equilibrium calculations fails actually results from an earlier call, for instance to TQINP with a phase name that doesn't exist.)
Log the ChemApp version number
If your program produes any kind of textual output, make sure it includes the ChemApp version number. This is especially useful if you store the output for documentation purposes: if you re-run the program later, potentially with another version of ChemApp, and discover differences in the output, you will be glad you stored this piece of information.
Before you contact GTT's support: Every output sent to GTT-Technologies' technical support should include the ChemApp version number, as returned by your program at run-time using TQVERS.
Make sure you are using the correct version of the ChemApp library
With almost all distributions of ChemApp, the ChemApp library comes in more than one version. All distributions contain at least a standard and an extended version. They differ in the maximum size of the chemical system they can handle. Make sure you don't use the standard version, when your data-file is too large for it and actually requires the extended version. See the file version.txt for details, it is part of your ChemApp distribution.
If you have been using a version of ChemApp light before you switched to the regular version, make sure you are not still using the "light" version.
Some distributions also include other versions of the ChemApp library. In case you are using a special version of ChemApp that allows you to add your own user models, make sure you don't use a regular version of the ChemApp library for linking instead.
If you are using a compiler-optimized version of ChemApp and you have doubts about the validity of your results, run your program with the non-optimized version too and see if the results are different. If they are, contact GTT-Technologies' technical support.
Before you contact GTT's support: If you have a problem with ChemApp and send your code to GTT-Technologies' technical support, make sure you have double-checked whether you are using the suitable library version. In case you are normally working with the standard version of ChemApp, make sure you have tested your program also with the extended version. In case you are normally working with a compiler-optimized version of ChemApp, make sure you first re-run your program using the non-optimized version of ChemApp, then check for differences.
Always check the error code every ChemApp subroutine returns
Every ChemApp subroutine has an error variable as its last parameter. Make sure your code checks this error variable after each call to a ChemApp subroutine. For examples on how to do this, see the cademo1 program. (Note that the short ChemApp example programs that are part of the reference section describing the various ChemApp subroutimes do purposely not follow this convention: to keep these code examples short and easy to read, error handling has usually been neglected.)
This is especially true when developing programs that have a graphical user interface (GUI): if you do not check the error variable after each call to a ChemApp subroutine, errors might go unnoticed until much later in your code, at which point they might result in an error which is misleading with respect to the reason why it occured.
The same is true for command line applications, if you use TQCIO to redirect your error messages to unit 0. This effectively hides the otherwise automatic reporting of errors.
Before you contact GTT's support: If you have a problem with ChemApp and send your code to GTT-Technologies' technical support, make sure your code has been written and run in a way so that any errors that ChemApp reports through this error variable were caught, reported, and didn't go unnoticed!
See also: TQERR.
Use ChemApp subroutines to open and close ChemApp data-files
Whenever possible, use ChemApp subroutines to open and close ChemApp data-files.
By selecting the proper ChemApp subroutine to open your ChemApp data-file which matches the format of your data-file (TQOPNA for ASCII data-files, TQOPNB for binary data-files, TQOPNT for transparent data-files), you make sure that your data-file is opened correctly.
Before you contact GTT's support: Make sure that the error you encountered is not related to the way you open the thermochemical data-file.
Make sure your ChemApp data-file exists and is loaded correctly
It is especially important to check the error variable when opening and reading ChemApp data-files.
Before you actually open a data-file, add appropriate code to your program that first makes sure this data-file really exists, and is found in the directory where you expect it.
Before you contact GTT's support: If you have a problem with ChemApp and send your code to GTT-Technologies' technical support, make sure you always add the data-file that you used when encountering the problem. Make sure the source of your problem is not the fact that you by accident try to load a data-file under a different name, or from another directory.
Make sure you close your data-file
Make sure that after opening and reading your data-file, you close it again. Otherwise problems might occur if you try to read the same data-file more than once, because then the file pointer is not located at the very beginning of the file.
Check the user id of data-files in transparent format
If you are using transparent data-files, ChemApp might refuse to load the data-file if certain conditions are not met. For instance, you might not be authorized to read the data-file because it was created with FactSage under a user id that doesn't match your ChemApp user id.
To catch these cases early on, have your ChemApp program report and log your user id (TQGTID) and optionally your user name (TQGTNM). After reading a transparent data-file successfully, consider reporting and logging information returned by TQGTRH.
Before you contact GTT's support: If you have a problem with reading a transparent data-file created with FactSage and send your code and transparent data-file to GTT-Technologies' technical support, make sure you understand the error message ChemApp reports. Double-check if the transparent data-file was created under a FactSage user id that matches your ChemApp user id.
Know your data and do not hard-wire index numbers
Don't try to save time by hard-wiring index numbers for system components, phases, phase constituents, and the like, unless you're still in the prototyping phase. Always let ChemApp tell you these index numbers based on the name of the entity that you're interested in. If your data-file ever gets changed, whether on purpose or by mistake, you might find yourself having to change these hard-wired portions of your code. The worst case is if you don't even notice that the data has changed, and you end up calculating something very different without realizing it.
For this reason, as well as for documentation purposes, you should consider having your program include a short table of contents of the data-file in your results or your log file. Have a look at worked example Print data-file contents in the ChemApp Programmer's Manual for suggestions.
Before you contact GTT's support: Make sure you do not use hard-wired index numbers in your code, but for instance always use the appropriate ChemApp subroutines to retrieve these numbers at run-time.
Archive your calculation results
If you want to or have to store results from your program for archival purposes (for instance by backing them up to tape or CD), don't forget to also store the thermochemical data-files you used to create them. From a ChemApp point of view, you should archive the following things:
- The results of your calculations, of course.
- All source code of your program (don't forget to include auxiliary files like Makefile, configuraton files, or other instructions on how to build the program).
- An executable version of your program. If you have both static and shared libraries/DLLs of ChemApp available, compile a statically linked version of your program.
- The thermochemical data-files used.
- The ChemApp library itself, especially when you're using ChemApp as a DLL or shared library.
Make use of ChemSage/ChemApp result tables
When writing customized programs based on ChemApp, especially those that either have a GUI, or those which are add-ons or modules for third-party programs, one is often tempted to exclusively rely on TQGETR to retrieve results from an equilibrium calculation.
While TQGETR gives you access to almost every piece of information you could want from an equilibrium calculation, it generally has one disadvantage: you will naturally only see the results you asked for. This can be a problem if you're still in the early stages of program development or of using a particular data-file, or when you are looking for reasons why your calculations don't give you the results you expected.
In these cases, a concise view of the whole equilibrium state is much more useful. This is exactly what a ChemSage/ChemApp result table provides, it gives you an overview of what your inputs were, what conditions you used, and what the equilibrium of your whole system looks like, all in an easy-to-read table.
Make use of these tables, which are produced when TQCEL, TQMAPL, or TQCENL are called. If your code has a GUI or is part of another program, consider sending this output to a log file. Consider using TQWSTR to add instructive comments to the same log file.
If you also have FactSage available, you might find it helpful to run a FactSage calculation with the same incoming amounts and conditions, since it gives you more interactive tools to explore a particular equilibrium calculation.
Before you contact GTT's support: If you have doubts about particular results you get from TQGETR, make sure you have also looked at the bigger picture, i.e. at other results that together describe the overall equilibrium state, for instance using the ChemSage/ChemApp result table output from subroutines like TQCEL.
Keep a watchful eye on your incoming amounts
In many programs, especially simpler ones, you likely know rather well what your incoming amounts are, because you often set the conditions for your equilibrium calculation yourself, or a user inputs them at run-time.
But there are types of programs where you don't necessarily always know what is input, and easily loose track of it:
- When your code is iterative, or contains a number of equilibrium calculations that are connected (i.e. incoming conditions for one equilibrium calculation are based on the results of an earlier one).
- When your code is part of another program, and the incoming conditions are supplied at run-time by other parts of the program, or are read from a file.
In both cases, you should provide means to make sure that the incoming conditions are still valid for the chemical system and the thermochemical data you are using. For instance, a temperature or an incoming amount might exceed an upper limit specified for the data you are using, thus potentially rendering the equilibrium results doubtful or even useless, or causing ChemApp not being able to calculate the equilibrium. The output from both TQSHOW and TQCEL, as well as calls to TQSETC with the "ia" option can help keeping track of incoming amounts.
Before you contact GTT's support: If you need help with an equilibrium results that looks doubtful, especially if your code is is iterative, make sure you carefully double-checked that the incoming amounts for this equilibrium calculation are what you intended them to be, and are valid with respect to the thermochemical data you are using.
Make sure you know which system units you are using
Suboutines used to define incoming amounts (e.g. TQSETC), as well as those used to retrieve results (e.g. TQGETR) only return the values, but not the associated units for temperature, pressure, energy, volume, or amount. The units used by ChemApp are always those which are currently active, and can be retrieved using TQGSU.
Don't automatically assume, especially in more complex programs, that you always know what the currently active system components are. Instead of hard-wiring the output of particular units, retrieve them at run-time using TQGSU.
As an example, do not hard-wire amount units in your output. You might think you're using the default value of "mol". But if you, somewhere in your code, retrieved molecular masses for phase constituents or stoichiometric compounds, you will likely have switched to unit "gram", in order to get the molecular masses in "gram/mol", which are generally expressed as "[current amount unit/mol]". If you forgot to switch back from "gram" after that, you might be confused by the results, if you hard-wired "mol" in your output. The same can happen if you hard-wire "gram/mol" in your output of the molecular masses, and are suprised that you get "1.0 gram/mol" for every phase constituents and stoichiometric compound. The reason is most likely that your amount unit is "mol", which gives you "mol/mol" as unit for the molecular masses. Retrieving the current amount unit at run-time to print the unit as "[current amount unit/mol]" makes this condition immediately obvious.
Facilitate debugging
In terms of ChemApp and what it calculates, good support for debugging basically means providing some features in your program that can help you look more closely at calculations that do not produce the results you expected. One way is to store the TQCEL output to a log file (see above).
If you really encounter problems with equilibrium calculations (ChemApp doesn't find an answer, ChemApp takes too long, etc.), you might not get an output from TQCEL, because the equilibrium calculation did not produce a result. In these cases, output from TQSHOW, called immediately before the equilibrium calculation with TQCE or TQCEL, can help tremendously. In fact, the output from TQSHOW, together with the thermochemical data-file you are using, and information about the target variable (if you are performing a target calculation) are the most helpful pieces of information for GTT-Technologies' technical support when trying to find the reasons for problems with individual equilibrium calculations that do not converge.
Instead of simply calling TQCE to calculate the equilibrium in your program, consider implementing the following code in order to quickly switch to "debug" mode, when need arises (this code is outlined in "pseudo code" and can be easily implemented in any programming language):
if DEBUGMODE NoOfEquilCalcs = NoOfEquilCalcs + 1 ErrorLog = "ERRLOG" + NoOfEquilCalcs + ".TXT" call TQCIO to redirect "LIST" output to ErrorLog call TQCIO to redirect "ERROR" output to ErrorLog open ErrorLog for writing call TQSHOW close ErrorLog open ErrorLog call TQCEL if NOERR from TQCEL is NOT zero call TQWSTR to store any additional useful information to ErrorLog close ErrorLog else close ErrorLog delete ErrorLog endif call TQCIO to redirect "LIST" and "ERROR" to their original units, if necessary else call TQCE endif
This simple code (which can still be improved) does the following things:
- If "DEBUGMODE" has been activated, it directs output to a dedicated log file (ErrorLog), which is given a unique name (in this example it is suggested that the number of equilibrium calculations performed this far is used to build a unique filename).
- It calls TQSHOW to log all current conditions (incoming amounts, pressure, temperature, etc.) to ErrorLog.
- It closes and reopens ErrorLog to make sure that this info is physically written to disk, in case your program hangs or crashes before TQCEL can produce output. This part of the code can be optimized, depending on which programming language you are using. For instance, instead of closing and reopening ErrorLog, you can also "flush" the write buffers of this file to disk, if the programming language you are using offers support for this.
- Only then is TQCEL called, which stores its output also to ErrorLog.
- If ChemApp has no problems calculating the equilibrium, the ErrorLog file is deleted. If TQCEL reports an error, ErrorLog contains all necessary information about the problematic equilibrium calculation. Note that even if your program hangs or crashes, ErrorLog still contains the TQSHOW output, which is often enough for GTT-Technologies' technical support to trace the problem, provided the thermochemical data-file is supplied too.
- If necessary (e.g. if other parts of your code depend on "ERROR" and "LIST" output being stored elsewhere), call TQCIO to restore the output destinations to their original values.
Before you contact GTT's support: If ChemApp encountered any problematic equilibrium calculations, all necessary information is thus stored to log files, one file for each problem. If you have problems with particular equilibrium calculations and would like to contact GTT-Technologies' technical support, this is what should be included with your email: The TQSHOW output and the section of the source code that performs the equilibrium calculations (always), the TQCEL output (if possible), and the data-file you used (always).
1.11 Thermodynamic data
All thermochemical data that ChemApp needs for its calculations are loaded from data-files, where each data-file contains the thermochemical data for a more or less complex chemical system.
A comprehensive list of readily available standard data-files that are adequate for many applications can be obtained from GTT-Technologies. Customised data-files can also be prepared to meet a user's specific requirements.
The data-files for ChemApp come in a number of forms: ASCII, transparent, and binary (the latter now being deprecated). It is necessary for every user of ChemApp (where the user is usually the programmer, or, depending on the application, the end user) to know which of these formats are used for his or her data-files. The reason is that each of these formats requires a slightly different code to be loaded into ChemApp. The details are described below (ASCII data-files, Binary data-files (DEPRECATED), and Transparent data-files).
A data-file contains the following information:
Components
- names of the default set
- molecular masses of the default set
Phases
- names
- list of constituents (one constituent in fixed composition phases)
- solution model parameters
- default status
Constituents
- names
- stoichiometries with respect to the default system components
- thermochemical data
- default status.
ChemApp can only read data stored in data-files in the various ChemApp formats (ASCII data-files, Binary data-files (DEPRECATED), and Transparent data-files). Data stored in a database or a database file must be read with a database management system (usually FactSage) which in turn produces ChemApp data-files.
Once the thermochemical data was loaded from the data-file, it is not possible to add phases or phase constituents to the chemical system. It is however possible to modify the thermochemical data, provided that the data-file loaded is in ASCII format (see Data Manipulation Subroutines). Also, the default system components, as well as the default status of phases and constituents may be changed (see Handling System Data in ChemApp), independent of the data-file format.
More information on the structure and handling of thermochemical data-files can be obtained from Thermochemical Data-files - Structure and Handling.
A significant feature of ChemApp is that it includes a comprehensive library of excess Gibbs energy models for various types of non-ideal solution phases. A list of these and typical areas for their application are summarised in Table List of available solution models in ChemApp. Additional 'customer-specified' models can be added upon request, and it is also possible for users to add their own solution models to ChemApp, as long as these models fulfill some specific requirements.
1.11.1 User-defined solution models
For users who develop their own solution models, a special version of ChemApp is available that might allow for the addition of these models. This would then be a three-step process, which can be summarised as follows:
- The user-defined solution model needs to be coded in Fortran and added to the skeleton subroutines USERGP, USERGX, and USERGG which are supplied together with the special version of ChemApp.
- In the second stage, this user code is compiled and either linked statically to the application program together with ChemApp, or made available as a DLL.
- The solution data need to be added to a thermochemical data-file.
If you are interested in adding your own solution models to ChemApp, please contact GTT-Technologies for further details.
| Model | Application area |
|---|---|
| For general use with substitutional or associated solution phases | |
| Solid phases with sublattice descriptions | |
|
Ionic liquids |
| Molten salts | |
| Ionic oxidic mixtures with or without non-oxidic solutes | |
| Condensed non-aqueous solutions | |
|
Binary condensed phases with a narrow stoichiometry range |
|
Metallic dilute solutions |
| Dilute aqueous solutions | |
| Concentrated aqueous solutions | |
|
Aqueous solutions up to 5 kbar and 1300K |
|
Fluid mixtures up to 1 Mbar and 6000K; important for many geological and environmental systems |
|
Non-ideal gas phases |
1.11.2 ASCII data-files
The ASCII (plain text) data-file format is the traditional format for thermochemical data-files for ChemApp. Every user of ChemApp has probably seen several of these, since each distribution of ChemApp contains example data-files in this format. See for instance below and Example data-files and their contents for an overview on the data-files in ASCII format included with ChemApp.
The most common and recommended file extension for thermochemical data-files in ASCII format is .DAT. ASCII data-files are portable among platforms, the only detail that needs to be observed is that DOS/Windows and Unix systems use different line endings for text files. The failure to do a proper conversion is one common reason for TQRFIL to report Error code 103.
| ASCII | Binary | Transparent | |
|---|---|---|---|
| How do I recognize it? | Plain text, can be viewed and edited with an ASCII editor | When viewed, does not contain readable data | When viewed, the first four readable characters of the file are 'CSTR' |
| Typical file extension | .DAT | .BIN | .CST |
| Minimum version of ChemApp required to read the file | V1.0.0 | FORTRAN: V2.1.4
Other: V3.3.0
|
V4.0.0 |
| Are the files portable across compilers and/or platforms? | Yes | No | Partially |
| Can I edit the data? | Yes, but the use of a program such as CSFAP is strongly recommended | No, do not attempt to edit, it will render the file useless | No, do not attempt to edit, it will render the file useless |
| How do I open it? | |||
| How do I read it? | TQRFIL | TQRBIN | TQRCST |
| How do I close it? | |||
| More information | ASCII data-files | Binary data-files (DEPRECATED) | Transparent data-files |
Using ASCII data-files (all programming languages)
The FORTRAN concept of unit numbers is not supported by other programming languages, thus the thermochemical data-files to be used by ChemApp cannot be opened with the standard file-opening subroutines of these languages. Even in cases where the application program is written in FORTRAN, it is still possible that the standard FORTRAN statements to open and close files (OPEN and CLOSE) cannot be used, for instance when using ChemApp in form of a DLL from a FORTRAN application program.
To both simplify and unify access to thermochemical data-files for all application programs and across all programming languages, a set of ChemApp subroutines is provided. This set of subroutines includes TQOPEN, TQOPNA, and TQCLOS.
The subroutine TQOPEN has been part of ChemApp since the earliest versions. It can both be used to open existing files for reading (e.g. thermochemical data-files), as well as to create new files for writing (e.g. files that are used to store the output of TQCEL, the ChemSage/ChemApp result tables). This flexibility, on the other hand, also comes with a disadvantage: If you try to open a data-file for reading, but the file is not where you or the program expects it to be, a new file of zero bytes length will be created instead. This effect is also mentioned in the description of TQOPEN.
Mainly because of this effect, another ChemApp subroutine has been added, which is available since version V3.3.0: TQOPNA. This subroutine opens the specified file only if it already exists. If it doesn't, or if any other error occurs during the file-opening process, it returns with Error code 107. As a note for users familiar with FORTRAN: TQOPNA opens the file with STATUS='OLD'.
It is thus recommended that TQOPNA be used instead of TQOPEN for opening thermochemical data-files which are in ASCII format. TQOPEN should be used when opening output files for writing, for instance for redirecting error messages, as well as for storing the output of TQSHOW, TQCEL, and TQMAPL to files (see TQCIO).
As of ChemApp V4.0.0, the subroutines TQOPEN, TQOPNA, and TQCLOS have been made part of the core code of ChemApp, and are thus available to all application languages, both FORTRAN and non-FORTRAN. We recommend using these subroutines also in FORTRAN application programs, instead of calling the standard FORTRAN statements OPEN and CLOSE, if you are using ChemApp version 4.0.0 or later.
Using FORTRAN as an example, a thermodynamic data-file in ASCII format could be opened with the following code:
C Open thermochemical data-file in ASCII format for reading
CALL TQOPNA('cosi.dat', 10, NOERR)
In case the attempt to open the data-file fails, TQOPNA will return with Error code 107, otherwise the data-file can now be loaded with TQRFIL and closed with TQCLOS:
C Read thermochemical data-file in ASCII format
CALL TQRFIL(NOERR)
C Close data-file
CALL TQCLOS(10, NOERR)
By default, TQRFIL expects the data-file to be opened with unit number 10, this being the reason for both the OPEN and CLOSE statements above referring to this particular unit number. It is also possible to use a non-default unit number for this purpose, in which case ChemApp needs to be told about this using TQCIO. ChemApp will accept unit numbers less then 11 and larger than 19. If ChemApp is part of a larger FORTRAN program, it is important to make sure that the unit number used to open the data-file is free, be it unit number 10 or a non-default value. The FORTRAN subroutine INQUIRE is helpful in this case.
This basic sequence of opening and loading a thermochemical data-file is used in virtually all example programs listed in this manual.
Using ASCII data-files (FORTRAN programs)
In FORTRAN terms, ASCII data-files are usually called text files or formatted sequential files and may be opened with the standard FORTRAN OPEN statement by the application program if this is written in FORTRAN and uses ChemApp in form of a static library. In case ChemApp is used as a DLL in conjunction with a FORTRAN application program, this might not work. For this reason the use of the subroutines TQOPNA and TQCLOS, as explained above, is recommended even for FORTRAN programs.
If the standard FORTRAN OPEN and CLOSE statements are to be used, the code segment dealing with loading an ASCII data-file might be similar to the following:
C Open thermochemical data-file in ASCII format for reading
OPEN(10, FILE='cosi.dat', STATUS='OLD', IOSTAT=NOERR)
IF (NOERR .NE. 0) THEN
WRITE(,) 'ERROR: Cannot open data-file.'
...
ENDIF
Note that the specifiers FORM= and ACCESS= can be omitted for ASCII data-files since their default values FORM='FORMATTED' and ACCESS='SEQUENTIAL' are the correct ones in this case.
Another point to note in the code example above is that if the OPEN statement fails, NOERR does not contain a ChemApp error number (see List of Error Messages), but one returned directly by the FORTRAN run time library. For details, please see your FORTRAN compiler's manual.
The syntax introduced above for opening a thermochemical data-file in ASCII format should be valid for all FORTRAN compilers on all platforms, as long as the data-file to be loaded has the correct line endings for text files on the platform in question. As already mentioned, this is an import detail to be aware of when transferring data-files between different computing platforms.
To make the process of opening data-files even safer, the use of an appropriate call to the FORTRAN statement INQUIRE prior to the OPEN statements is recommended.
The opened file can then subsequently be loaded into ChemApp with TQRFIL, and closed with CLOSE:
C Read thermochemical data-file in ASCII format
CALL TQRFIL(NOERR)
IF (NOERR .NE. 0) THEN
WRITE(,) 'ERROR loading data-file.'
...
ENDIF
C Close data-file
CLOSE(10)
In terms of the FORTRAN unit number, the same notes apply as above: By default, TQRFIL expects the data-file to be opened with unit number 10, this being the reason for both the OPEN and CLOSE statements above referring to this particular unit number. It is also possible to use a non-default unit number for this purpose, in which case ChemApp needs to be told about this using TQCIO. ChemApp will accept unit numbers less then 11 and larger than 19. If ChemApp is part of a larger program, it is important to make sure that the unit number used to open the data-file is free, be it unit number 10 or a non-default value. Again, the FORTRAN subroutine INQUIRE is helpful in this case.
1.11.3 Binary data-files (DEPRECATED)
| Note: | The binary data-file format is a legacy format that has been superceded by the transparent file format (see Transparent data-files below). If you still have files in this format, please contact GTT-Technologies to help you convert these files to the transparent file format, if necessary. |
|---|
Binary thermochemical data-files for use with ChemApp are stored in a way which does not permit the end user to extract or change thermochemical data from the file. This is mainly done in cases where a license agreement with the supplier of the data does not permit the distribution in plain text format.
Due to the nature of the FORTRAN standard for writing binary data to files, or rather, the lack thereof, binary data-files are usually not portable across platforms, and in many case are not even portable between different compilers on the same platform (this problem is partially solved by using data-files in transparent format, see below). For the suppliers of binary data-files (in most cases this is GTT-Technologies), this means that the data-file has to be supplied for the very compiler and/or platform that the ChemApp customer uses. For the ChemApp user it means that care has to taken that the binary data-file supplied is only used with the proper compiler. If you try to load a binary data-file into ChemApp that was actually made for a different compiler, you typically will receive Error code 103 from TQRBIN.
The most common and recommended file extension for thermochemical data-files in binary format is .BIN. They are otherwise recognized by the fact that when inspected with a file-viewer, their contents are not humanly readable. It is important to realise that a binary data-file should never be edited manually, as it will render the file useless.
Due to the lack of portability of binary files across platforms and compilers, the support of thermochemical data-files in binary format will be discontinued in future versions of ChemApp. As of ChemApp version 4.0.0 the binary file format is superceded by the transparent file format (see Transparent data-files). If you are using ChemApp versions 4.0.0 and later with binary data-files, please contact GTT-Technologies to receive help in converting these data-files to the transparent format.
Using binary data-files (all programming languages)
For the same reasons as mentioned above (see ASCII data-files), a set of subroutines is provided to open and close binary data-files from FORTRAN and non-FORTRAN programs. These subroutines are TQOPNB and TQCLOS. It is important to realise that TQOPEN cannot be used to open binary data-files, as it will always open the data-file specified in ASCII mode. This of course does not affect the use of TQOPEN to create new ASCII files for writing, for instance for redirecting error messages, as well as for storing the output of TQSHOW, TQCEL, and TQMAPL to files (see TQCIO).
As of ChemApp V4.0.0, the subroutines TQOPNB, and TQCLOS have been made part of the core code of ChemApp, and are thus available to all application languages, both FORTRAN and non-FORTRAN. We recommend using these subroutines also in FORTRAN application programs instead of calling the standard FORTRAN statements OPEN and CLOSE, if you are using ChemApp version 4.0.0 or later.
Using FORTRAN as an example, a thermodynamic data-file in ASCII format could be opened with the following code:
C Open thermochemical data-file in binary format for reading
CALL TQOPNB('cosi.bin', 10, NOERR)
In case the opening of the data-file fails, TQOPNB will return with Error code 107, otherwise the data-file can now be loaded with TQRBIN and closed with TQCLOS:
C Read thermochemical data-file in binary format
CALL TQRBIN(NOERR)
C Close data-file
CALL TQCLOS(10, NOERR)
Note especially that for binary data-files, TQRBIN needs to be used to load the data-file, not TQRFIL or TQRCST.
With respect to the unit numbers, please see the previous section (Chapter ASCII data-files) for explanatory notes, they apply equally to binary data-files.
Using binary data-files (FORTRAN programs)
In FORTRAN terms, binary data-files are usually called unformatted sequential files, and are opened with the standard FORTRAN OPEN statement, for instance with a sequence similar to the following:
C Open thermochemical data-file in binary format for reading
OPEN(10, FILE='cosi.bin', STATUS='OLD', FORM='UNFORMATTED',
* IOSTAT=NOERR)
IF (NOERR .NE. 0) THEN
WRITE(,) 'ERROR: Cannot open data-file.'
...
ENDIF
Note that the specifier ACCESS= can be omitted, since its default value ACCESS='SEQUENTIAL' is the correct one in this case.
Another point to note in the code example above is that if the OPEN statement fails, NOERR does not contain a ChemApp error number (see List of Error Messages), but one returned directly by the FORTRAN run time library. For details, please see your FORTRAN compiler's manual.
The syntax introduced above for opening thermochemical data-files in binary format should be valid for all FORTRAN compilers on all platforms, but it is important to realise that the binary data-files themselves are usually not portable across platforms, and in many case not even among different compilers on the same platform.
To make the process of opening data-files even safer, the use of an appropriate call to the FORTRAN subroutine INQUIRE prior to the OPEN statement is recommended.
The opened file can then subsequently be loaded into ChemApp with TQRBIN, and closed with CLOSE:
C Read thermochemical data-file in binary format
CALL TQRBIN(NOERR)
IF (NOERR .NE. 0) THEN
WRITE(,) 'ERROR loading data-file.'
...
ENDIF
C Close data-file
CLOSE(10)
Note especially that for binary data-files, TQRBIN needs to be used to load the data-file, not TQRFIL or TQRCST.
With respect to the unit numbers, please see the previous section (ASCII data-files) for explanatory notes, they apply equally to binary data-files.
1.11.4 Transparent data-files
A drawback of the FORTRAN language is the lack of a standard for binary files. This is the main reason why ChemApp data-files in binary format are not portable (see above). To at least partially overcome this problem, the transparent data-file format has been created. Using transparent data-files, approximately 90% of the portability problems on Windows platforms can be overcome.
Transparent data-files differ from data-files in ASCII and binary format in that they have a header which precedes the actual thermochemical data, and which for instance contains information about the data itself, as well as the creator and user of the data-file (see TQGTRH).
On systems running Microsoft Windows, transparent data-files can be used by programs which contain a version of ChemApp that was compiled with any of the following compilers:
- Lahey FORTRAN
- Microsoft PowerStation FORTRAN
- Digital Visual FORTRAN
- Compaq Visual FORTRAN
- Intel Visual FORTRAN
Since these compilers are also used to produce FactSage, and all ChemApp DLLs for the Windows platforms (ChemApp DLLs for use with Microsoft Visual Basic and Visual C++, as well as Borland Delphi), this for instance enables a transparent data-file written by FactSage to be read by ChemSheet without modification.
While transparent data-files can be created by GTT-Technologies for every platform for which ChemApp is available, one thing to remember is that transparent data-files created for the Windows-based compilers listed above will very likely not be directly usable on a Unix system, for instance. If you attempt to read a transparent data-file under such a condition, you will typically get Error code 161. These portability issues will be solved with the future portable data-file format.
Transparent data-files, like binary data-files, are stored in a way that does not permit the end user to extract or change thermochemical data from the data-file. This is mainly done in cases where a license agreement with the supplier of the data does not permit the distribution in plain text format.
The most common and recommended file extension for thermochemical data-files in transparent format is .CST. They are otherwise recognized by the fact that when inspected with a file-viewer, their contents are not humanly readable, apart from a comment in the file header: This is a ChemSage transparent data file... . It is important to realise that a transparent data-file should never be edited manually, since ChemApp will refuse to load a modified transparent data-file.
On Windows systems, transparent data-files created for use with any of the compilers listed above can also be identified by examining the very first four characters, the magic bytes, of the file, which are always CSTR.
Using transparent data-files (all programming languages)
As with data-files in ASCII (see ASCII data-files) and binary (see Binary data-files (DEPRECATED)) formats, the use of a set of subroutines is provided to open and close transparent data-files from FORTRAN and non-FORTRAN programs. These subroutines are TQOPNT and TQCLOS. It is important to realise that TQOPEN cannot be used to open transparent data-files, as it will always open the data-file specified in ASCII mode. This of course does not affect the use of TQOPEN to create new ASCII files for redirecting error messages, as well as for storing the output of TQSHOW, TQCEL, and TQMAPL to files (see TQCIO).
Using FORTRAN as an example, opening a thermodynamic data-file in transparent format would thus be similar to the following piece of code:
C Open thermochemical data-file in transparent format for reading
CALL TQOPNT('cosi.cst', 10, NOERR)
In case the opening of the data-file fails, TQOPNT will return with Error code 107, otherwise the data-file can now be loaded with TQRCST and closed with TQCLOS:
C Read thermochemical data-file in transparent format
CALL TQRCST(NOERR)
C Close data-file
CALL TQCLOS(10, NOERR)
Note especially that for transparent data-files, TQRCST needs to be used to load the data-file, not TQRFIL or TQRBIN.
With respect to the unit numbers, please see the previous section (ASCII data-files) for explanatory notes, they apply equally to transparent data-files.
Using transparent data-files (FORTRAN programs)
In FORTRAN programs, transparent data-files can be both opened using the ChemApp subroutine TQOPNT as described above, as well as using the standard FORTRAN OPEN statement. While the use of the FORTRAN statement OPEN may be more flexible for this purpose, its calling syntax when used to open a transparent data-file is compiler-dependent. Note also that as with opening data-files in ASCII and binary format, using FORTRAN's OPEN may not work if you are using ChemApp in the form of a DLL. Unless you need the added flexibility of the OPEN statement, we thus recommend using TQOPNT as described above.
Here are some examples of how to open a transparent data-file using the OPEN statement on various compilers. See below for a compiler-independent version that uses TQOPNT.
Note that if any of the OPEN statement fails in the examples below, the error code returned in NOERR does not contain a ChemApp error number (see List of Error Messages), but an error code returned directly by the FORTRAN run time library. For details, please see your FORTRAN compiler's manual.
To make the process of opening data-files even safer, the use of an appropriate call to the FORTRAN statement INQUIRE prior to the OPEN statement is recommended.
Lahey FORTRAN
Under Lahey FORTRAN, the specifier ACCESS='TRANSPARENT' is used to open ChemApp transparent data-files, hence we named these files transparent data-files.
C Open thermochemical data-file in transparent format for reading
C (Lahey version)
OPEN(10,FILE='cosi.cst', STATUS='OLD', ACCESS='TRANSPARENT',
* IOSTAT=NOERR)
IF (NOERR .NE. 0) THEN
WRITE(,) 'ERROR: Cannot open data-file.'
...
ENDIF
Note that there is no FORM='...' specifier used.
The data-file is then subsequently read using TQRCST and closed using CLOSE (see below).
Unix FORTRAN and other FORTRAN compilers
The ACCESS='TRANSPARENT' and FORM='BINARY' specifiers described above are not standard FORTRAN, so many compilers do not implement them. This is the case for many, if not all, FORTRAN compilers on Unix systems, and includes for instance the Gnu g77 FORTRAN compiler on Linux systems. You thus cannot use transparent data-files with these non-Windows compilers, unless they have been specially created for use with them. In most cases this is done by GTT-Technologies.
If you were supplied with such a data-file, open it using FORM='UNFORMATTED', just like a binary data-file:
C Open thermochemical data-file in transparent format for reading
C (Version for Unix FORTRAN and other FORTRAN compilers, e.g. g77)
OPEN(10, FILE='cosi.cst', STATUS='OLD', FORM='UNFORMATTED',
* IOSTAT=NOERR)
IF (NOERR .NE. 0) THEN
WRITE(,) 'ERROR: Cannot open data-file.'
...
ENDIF
The data-file is then subsequently read using TQRCST and closed using CLOSE (see below).
All FORTRAN compilers: TQOPNT
If you need your FORTRAN programs to compile under various compilers and/or platforms, and do not need the added flexibility of the generic FORTRAN OPEN statement, you avoid any compiler-dependent code by using the ChemApp subroutine TQOPNT.
C Open thermochemical data-file in transparent format for reading
C (Version for all FORTRAN compilers using TQOPNT)
CALL TQOPNT('cosi.cst', 10, NOERR)
IF (NOERR .NE. 0) THEN
WRITE(,) 'ERROR: Cannot open data-file.'
STOP
ENDIF
Again, the syntax introduced above for opening thermochemical data-files in transparent format using TQOPNT is valid for all FORTRAN compilers on all platforms, but it is important to realise that the transparent data-files themselves are usually not fully portable across platforms, and in many case not even among different compilers on the same platform, with the notable exception of the Lahey FORTRAN, Microsoft PowerStation FORTRAN, Digital Visual FORTRAN, and Compaq Visual FORTRAN compilers on Windows platforms.
With the respect to the unit numbers, please see the previous chapter (ASCII data-files) for explanatory notes, they apply equally to transparent data-files.
The opened transparent data-file can then subsequently be loaded into ChemApp with TQRCST, and closed with TQCLOS:
C Read thermochemical data-file in transparent format
CALL TQRCST(NOERR)
IF (NOERR .NE. 0) THEN
WRITE(,) 'ERROR loading data-file.'
...
ENDIF
C Close data-file
CALL TQCLOS(10, NOERR)
IF (NOERR .NE. 0) THEN
WRITE(,) 'ERROR: Cannot close data-file.'
STOP
ENDIF
Note especially that for transparent data-files, TQRCST needs to be used to load the data-file, not TQRFIL or TQRBIN.
1.12 Example thermochemical data-files
ChemApp comes with a set of example data-files. This should enable you to start programming with ChemApp right away, even if you do not already have specific thermochemical data-files for your particular application. Some of the example data-files included in the ChemApp distribution are also used by the example programs which are part of this documentation. Below you find a description of all the example data-files included, together with their potential applications, as well as relevant temperature and composition application ranges.
1.12.1 COSI.DAT (System C-O-Si)
| Chemical system: | |
|---|---|
| C-O-Si | |
| Potential applications: | |
| Phase equilibria and thermodynamic properties, for instance relating to Si production in a Si-arc furnace. | |
| Temperature range: | |
| RT - 3500K | |
| Composition range: | |
| All | |
| Comments: | COSI.DAT is the file most widely used for the code examples in this manual. It contains a gaseous mixture phase and several pure stoichiometric phases. |
1.12.2 CNO.DAT (System C-N-O)
| Chemical system: | |
|---|---|
| C-N-O | |
| Potential applications: | |
| Combustion equilibria and thermodynamic properties of the gas species and condensed phases. | |
| Temperature range: | |
| All | |
| Composition range: | |
| All | |
| Comments: | This data-file contains 28 gas constituents as well as solid carbon and diamond. |
1.12.3 FEC.DAT (System Fe-C)
| Chemical system: | |
|---|---|
| Fe-C | |
| Potential applications: | |
| Phase equilibria; phase diagram, also metastable with cementite; thermodynamic properties of the phases and their constituents. | |
| Temperature range: | |
| RT - 2500K | |
| Composition range: | |
| All | |
| Comments: | See comment section at the end of the data-file (FEC.DAT data-file ) |
1.12.4 FEMGSIO4.DAT (System Fe2SiO4-Mg2SiO4)
| Chemical system: | |
|---|---|
| System Fe2SiO4-Mg2SiO4 | |
| Potential applications: | |
| Solid T-P-composition equilibria and thermal properties in the system Fe2SiO4-Mg2SiO4 for pressures between 1 bar and 300 kbar. | |
| Temperature range: | |
| Below liquidus | |
| Composition range: | |
| All | |
| Comments: | This data-file contains pressure-dependent Gibbs energy terms. |
1.12.5 PBSN.DAT (System Pb-Sn)
| Chemical system: | |
|---|---|
| Pb-Sn | |
| Potential applications: | |
| Phase equilibria and all thermodynamic properties in the binary system Pb-Sn. | |
| Temperature range: | |
| RT - 1500 K | |
| Composition range: | |
| All | |
| Comments: | This data-file can be used to calculate the Pb-Sn binary phase diagram, as it is valid over the whole composition range and contains the descriptions of all necessary condensed phases. Due to the inherent metastable miscibility gap, the BCT_A5 phase (Sn-terminal solution) is present twice. |
1.12.6 PITZER.DAT (System Na-Cl-C-O-H)
| Chemical system: | |
|---|---|
| Na-Cl-C-O-H | |
| Potential applications: | |
| Chemical equilibria in natural waters with sodium. | |
| Temperature range: | |
| 298.15 K | |
| Composition range: | |
| Aqueous solutions with up to 10 molal dissolved salts. | |
| Comments: | This data-file contains an aqueous phase modelled according to the Pitzer formalism. It contains charged species in the aqueous phase, which is the reason why this data-file actually has a sixth system component, the electron. |
1.12.7 WATER.DAT (System O-H-EA)
| Chemical system: | |
|---|---|
| O-H-EA | |
| Potential applications: | |
| Gas-Liquid-Solid equilibria of water with non-ideal gas and liquid with phase internal equilibria. | |
| Temperature range: | |
| From approx. 270 K to about 3000 K | |
| Composition range: | |
| Aqueous phase with gas and solid. | |
| Comments: | This file serves as an example of input for non-ideal gas using Tsonopoulos data (Tc,Pc,Vc, acentric factor and dipol moment) as well as Ideal Water solution with neutral and ionic species. |
1.12.8 SUBL-EX.DAT (System Co-Cr-Fe)
| Chemical system: | |
|---|---|
Co-Cr-Fe (incomplete) |
|
| Potential applications: | |
SIGMA-BCC equilibria |
|
| Temperature range: | |
900 - 1200 K |
|
| Composition range: | |
Iron-rich or chromium-rich. |
|
| Comments: | This file serves as an example of input for substitutional solutions (BCC) described with the RKMP model (see Table 2) and for a multi-sublattice phase (SIGMA) with three sublattices and 30 atoms of the unit cell. Please note that this sample data-file is not a comprehensive data-file for the system Co-Cr-Fe. Phases such as FCC and HCP have been omitted to keep it short. |
2 Initialising the Interface and Reading a Data-file
The first steps when programming ChemApp are
- to initialise the interface; this mainly causes ChemApp to set default values for temperature and pressure
- to read a pre-prepared ChemApp data-file.
Initialisation of the interface is done with a call to TQINI. The subroutine TQRFIL enables a thermodynamic data-file in ASCII format to be read. In case you have data-files supplied in binary or transparent format, use TQRBIN or TQRCST, respectively, instead. After loading a transparent data-file, TQGTRH retrieves information from the transparent file header.
TQCPRT retrieves a copyright message for ChemApp, while TQVERS gets its version number. TQLITE can be used to determine whether the ChemApp library used is ChemApp light or the regular version of ChemApp. TQSIZE retrieves a number of parameters which indicate the sizes of a number of internal arrays of the version of ChemApp used, while TQUSED returns a similar set of values for the currently loaded thermochemical system.
The subroutines TQCIO and/or TQCSU need to be called if any default settings relating to input/output or system units are to be modified. Similarly, TQGIO and TQGSU are used to determine the current values of these options.
TQOPNA, TQOPNB, TQOPNT, TQCLOS, and TQOPEN make sure that when programming in a language other than FORTRAN, files used by ChemApp are opened and closed using the correct FORTRAN unit numbers. Even when programming in FORTRAN, the use of these subroutines over FORTRAN's OPEN and CLOSE is still strongly recommended.
When opening an ASCII output file for writing with TQOPEN, for instance to store ChemSage/ChemApp result tables, TQWSTR can be used to write user-defined text to the same file.
As of Version 4.0.0, each ChemApp library contains information on the license holder (see TQGTID and TQGTNM), and can be identified via a short signature string (see TQGTPI).
TQCONF was added in version 6.3.4 to allow certain configuration options to be set.
All the subroutines used in these procedures are summarised in Table ChemApp subroutines for the initialisation stage, and described in more detail below.
| Subroutine | Function |
|---|---|
| TQINI | Initialises ChemApp |
| TQCPRT | Gets copyright message |
| TQVERS | Gets the ChemApp version number |
| TQLITE | Checks whether ChemApp light is used |
| TQGTID | Gets the user ID of the license holder of ChemApp |
| TQGTNM | Gets the name of the license holder of ChemApp |
| TQGTPI | Gets the ID of the program |
| TQGTHI | Gets the HASP dongle type and id |
| TQGTED | Gets the expiration date of the ChemApp license |
| TQCONF | Sets a configuration option |
| TQSIZE | Gets the internal array dimensions of ChemApp |
| TQUSED | Gets the dimensions of the currently loaded thermochemical system |
| TQGIO | Gets the value of an output option |
| TQCIO | Changes the value of an output option |
| TQRFIL | Reads a thermodynamic data-file in ASCII format |
| TQRBIN | Reads a thermodynamic data-file in binary format |
| TQRCST | Reads a thermodynamic data-file in transparent format |
| TQOPEN | Opens a file |
| TQWSTR | Writes a character string to the units associated with 'LIST' or 'ERROR' |
| TQOPNA | Opens a thermochemical data-file in ASCII format |
| TQOPNB | Opens a thermochemical data-file in binary format |
| TQOPNT | Opens a thermochemical data-file in transparent format |
| TQCLOS | Closes a file |
| TQGTRH | Retrieves information stored in the header of a transparent file |
| TQGSU | Gets a system unit |
| TQCSU | Changes a system unit |
| Subroutine | Function |
|---|---|
| Temperature | 1000 K |
| Pressure | 1 bar |
| Composition | No incoming amounts, no streams |
| System units | bar, dm3, Kelvin, Joule, mol |
2.1 TQINI
INITIALISE-INTERFACE
Use TQINI to initialise ChemApp.
Synopsis
- FORTRAN:
- CALL TQINI(NOERR)
- C:
- tqini(&noerr);
- Pascal:
- tqini(noerr);
- Basic:
- Call tqini(noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NOERR | INTEGER | Returns an error number |
TQINI must be called before any other ChemApp subroutine, or whenever you wish to reinstate the complete set of default values and units (see Table 4), and read a thermodynamic data-file.
- See also
- TQREMC
! The shortest possible (but very useless) ChemApp program PROGRAM CAF1 IMPLICIT NONE INTEGER NOERR ! Initialise ChemApp CALL TQINI(NOERR) END
/* Program cac1 */ /* The shortest possible (but very useless) ChemApp program */ #include "cacint.h" int main() { LI noerr; /* Initialise ChemApp */ tqini(&noerr); return 0; }
2.2 TQCPRT
GET-COPYRIGHT-MESSAGE
Use TQCPRT to get the copyright message for ChemApp
Added for ChemApp version 1.1.3
Synopsis
- FORTRAN:
- CALL TQCPRT(NOERR)
- C:
- tqcprt(&noerr);
- Pascal:
- tqcprt(noerr);
- Basic:
- Call tqcprt(noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NOERR | INTEGER | Returns an error number |
TQCPRT provides the programmer with a copyright message for inclusion in application programs utilising ChemApp. The message itself is retrieved like an error message, with a subsequent call to TQERR.
- See also
- TQERR
! Retrieving and displaying the ChemApp copyright notice PROGRAM CAF2 IMPLICIT NONE INTEGER NOERR CHARACTER MESS(3)*80 ! Initialise ChemApp CALL TQINI(NOERR) ! Retrieve the copyright string with TQCPRT CALL TQCPRT(NOERR) ! Then call tqerr to retrieve it CALL TQERR(MESS,NOERR) ! To print out the message, print all three strings of MESS : WRITE(UNIT=*,FMT='(3(A,/))') MESS(1), MESS(2), MESS(3) END
This program contains ChemApp Copyright GTT-Technologies, Kaiserstrasse 100, D-52134 Herzogenrath, Germany http://www.gtt-technologies.de
/* Program cac2 */ /* Retrieving and displaying the ChemApp copyright notice */ #include "cacint.h" int main() { LI noerr; int i; /* Initialise ChemApp */ tqini(&noerr); /* Retrieve the copyright string with tqcprt */ tqcprt(&noerr); /* Then call tqerr to retrieve it. The variable TQERRMSG is declared in cacint.c as TQERRMSG[3][80] for this purpose:*/ tqerr((CHP)TQERRMSG,&noerr); /* Note that an explicit typecast has been used in the above call to tqerr to avoid potential compiler warnings about TQERRMSG being an incompatible pointer type */ /* To print out the message, loop over the three strings of TQERRMSG: */ for(i=0;i<3;i++) printf("%s\n",TQERRMSG[i]); return 0; }
This program contains ChemApp Copyright GTT-Technologies, Kaiserstrasse 100, D-52134 Herzogenrath, Germany http://www.gtt-technologies.de
2.3 TQVERS
GET-VERSION-NUMBER
Use TQVERS to get the version number of ChemApp
Added for ChemApp version 1.1.3
Synopsis
- FORTRAN:
- CALL TQVERS(NVERS,NOERR)
- C:
- tqvers(&nvers,&noerr);
- Pascal:
- tqvers(nvers,noerr);
- Basic:
- Call tqvers(nvers,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NVERS | INTEGER | Returns the version number of ChemApp |
| NOERR | INTEGER | Returns an error number |
NVERS is returned as an integer value, e.g. 213 for version 2.1.3 of ChemApp.
! Display the version number of ChemApp PROGRAM CAF3 IMPLICIT NONE INTEGER NOERR, NVERS ! Initialise ChemApp CALL TQINI(NOERR) ! Get version number of ChemApp CALL TQVERS(NVERS,NOERR) WRITE(UNIT=*,FMT='(A,I4)') 'This program has been compiled '// * 'with ChemApp version ', NVERS END
This program has been compiled with ChemApp version 645
/* Program cac3 */ /* Display the version number of ChemApp */ #include "cacint.h" int main() { LI noerr, version; /* Initialise ChemApp */ tqini(&noerr); /* Get version number of ChemApp*/ tqvers(&version,&noerr); printf("This program has been compiled " "with ChemApp version %li\n", version); return 0; }
This program has been compiled with ChemApp version 645
2.4 TQLITE
CHECK-IF-CHEMAPP-LIGHT
Use TQLITE to check whether the type of ChemApp currently used is ChemApp light.
Added for ChemApp version 2.0.1
Synopsis
- FORTRAN:
- CALL TQLITE(ISLITE,NOERR)
- C:
- tqlite(&islite,&noerr);
- Pascal:
- tqlite(islite,noerr);
- Basic:
- Call tqlite(islite,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| ISLITE | INTEGER | Returns 1 for ChemApp light, 0 for ChemApp |
| NOERR | INTEGER | Returns an error number |
TQLITE can be used to distinguish between the regular version of the ChemApp library, and ChemApp light.
ChemApp and ChemApp light differ in a couple of ways (see About ChemApp and ChemApp light). If TQLITE returns a 0 through ISLITE, the regular version of ChemApp is linked to the calling program. If a 1 is returned, ChemApp light is used.
The code examples of the manual make use of this subroutine primarily to determine whether target calculations can be performed.
! Testing for the presence of ChemApp "light" PROGRAM CAF23 IMPLICIT NONE INTEGER NOERR, ISLITE ! Initialise ChemApp CALL TQINI(NOERR) 20 FORMAT(1X,A) ! Find out whether we are working with the regular or the "light" ! version CALL TQLITE(ISLITE, NOERR) WRITE(*,FMT=20) * 'This application program has been linked against' IF (ISLITE .EQ. 1) THEN WRITE(*,FMT=20) * 'ChemApp "light".' ELSE WRITE(*,FMT=20) * 'the regular version of ChemApp.' ENDIF END
This application program has been linked against the regular version of ChemApp.
/* Program cac23 */ /* Testing for the presence of ChemApp "light" */ #include "cacint.h" int main() { LI noerr, islite; /* Initialise ChemApp */ tqini(&noerr); /* Find out whether we are working with the regular or the "light" version */ tqlite(&islite, &noerr); printf("This application program has been linked against\n"); if (islite) printf("ChemApp \"light\".\n"); else printf("the regular version of ChemApp.\n"); return 0; }
This application program has been linked against the regular version of ChemApp.
2.5 TQGTID
GET-USER-ID
Use TQGTID to get the user ID of the license holder of the ChemApp library.
Added for ChemApp version 4.0.0
Synopsis
- FORTRAN:
- CALL TQGTID(ID,NOERR)
- C:
- tqgtid(id,&noerr);
- Pascal:
- tqgtid(id,noerr);
- Basic:
- Call tqgtid(id,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| ID | CHARACTER | Returns the user ID (255 chars) |
| NOERR | INTEGER | Returns an error number |
TQGTID returns the user ID of the license holder of ChemApp.
Note that the user ID which is returned can be up to 255 characters long, the variable ID should thus be declared large enough to hold a character string of this length.
! Retrieving and displaying the user ID and the name of the ChemApp ! license holder, the program ID, HASP dongle information, plus the ! expiration date of the ChemApp license. ! If the ChemApp version used requires a HASP dongle, and the ChemApp ! license is expired, or about to expire, and is to be extended, the ! information printed by this program is needed by GTT-Technologies to ! update the dongle. ! The support function STRLEN returns the total number of characters in ! a string, not counting trailing blanks INTEGER FUNCTION STRLEN(INSTR) CHARACTER INSTR*(*) STRLEN = LEN(INSTR) DO WHILE(INSTR(STRLEN:STRLEN) .EQ. ' ') STRLEN = STRLEN - 1 IF (STRLEN .EQ. 1) RETURN ENDDO RETURN END PROGRAM CAF30 IMPLICIT NONE INTEGER NOERR, STRLEN, HASPID, EDMON, EDYEAR CHARACTER ID*255, NAME*80, PID*24, HASPT*5 ! Initialise ChemApp CALL TQINI(NOERR) ! Retrieve the licensee's user ID CALL TQGTID(ID,NOERR) ! Retrieve the licensee's name CALL TQGTNM(NAME,NOERR) ! Retrieve the program ID CALL TQGTPI(PID,NOERR) ! Print all three WRITE(UNIT=*,FMT=*) 'Licensee''s user ID: ', ID(1:STRLEN(ID)) WRITE(UNIT=*,FMT=*) 'Licensee''s name : ', NAME(1:STRLEN(NAME)) WRITE(UNIT=*,FMT=*) 'Program ID : ', PID(1:STRLEN(PID)) ! The following pieces of information are only meaningful if a version ! of ChemApp is used that requires a dongle (hardware key). ! Get the HASP dongle type and id CALL TQGTHI(HASPT, HASPID, NOERR) ! Get the ChemApp license expiration date (month and year) CALL TQGTED(EDMON, EDYEAR, NOERR) ! Print info if HASP dongle is used: IF (HASPID .NE. 0) THEN WRITE(UNIT=*,FMT=*) 'HASP dongle type : ' // * HASPT(1:STRLEN(HASPT)) WRITE(UNIT=*,FMT=*) 'HASP dongle id : ', HASPID WRITE(UNIT=*,FMT=*) 'ChemApp license expiration date ' // * '(month/year): ', EDMON, '/', EDYEAR ELSE WRITE(UNIT=*,FMT=*) 'This ChemApp version does not ' // * 'require a HASP hardware key (dongle)' ENDIF END
Licensee's user ID: 5001 Licensee's name : GTT - Technologies Program ID : CAFU This ChemApp version does not require a HASP hardware key (dongle)
/* Program cac30 */ /* Retrieving and displaying the user ID and the name of the ChemApp license holder, as well as the program ID */ /* If the ChemApp version used requires a HASP dongle, and the ChemApp license is expired, or about to expire, and is to be extended, the information printed by this program is needed by GTT-Technologies to update the dongle. */ #include "cacint.h" int main() { LI noerr, haspid, edmon, edyear; char id[255], name[80], pid[TQSTRLEN], haspt[TQSTRLEN]; /* Initialise ChemApp */ tqini(&noerr); /* Retrieve the licensee's user ID */ tqgtid(id, &noerr); /* Retrieve the licensee's name */ tqgtnm(name, &noerr); /* Retrieve the program ID */ tqgtpi(pid, &noerr); /* Print all three */ printf("Licensee's user ID: %s\n", id); printf("Licensee's name : %s\n", name); printf("Program ID : %s\n", pid); /* The following pieces of information are only meaningful if a version of ChemApp is used that requires a dongle (hardware key). Get the HASP dongle type and id */ tqgthi(haspt, &haspid, &noerr); /* Get the ChemApp license expiration date (month and year) */ tqgted(&edmon, &edyear, &noerr); /* Print info if HASP dongle is used: */ if (haspid) { printf("HASP dongle type : %s\n", haspt); printf("HASP dongle id : %li\n", haspid); printf("ChemApp license expiration date (month/year): %li/%li\n", edmon, edyear); } else { printf("This ChemApp version does not require a " "HASP hardware key (dongle)\n"); } return 0; }
Licensee's user ID: 5001 Licensee's name : GTT - Technologies Program ID : CAFU This ChemApp version does not require a HASP hardware key (dongle)
2.6 TQGTNM
GET-USER-NAME
Use TQGTNM to get the name of the license holder of the ChemApp library.
Added for ChemApp version 4.0.0
Synopsis
- FORTRAN:
- CALL TQGTNM(NAME,NOERR)
- C:
- tqgtnm(name,&noerr);
- Pascal:
- tqgtnm(name,noerr);
- Basic:
- Call tqgtnm(name,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NAME | CHARACTER | Returns the name (80 chars) |
| NOERR | INTEGER | Returns an error number |
TQGTNM returns the name of the license holder of ChemApp.
Note that the name which is returned can be up to 80 characters long, the variable NAME should thus be declared large enough to hold a character string of this length.
- See
- TQGTID
2.7 TQGTPI
GET-PROGRAM-ID
Use TQGTPI to get the ID of the program.
Added for ChemApp version 4.0.0
Synopsis
- FORTRAN:
- CALL TQGTPI(NAME,NOERR)
- C:
- tqgtpi(name,&noerr);
- Pascal:
- tqgtpi(name,noerr);
- Basic:
- Call tqgtpi(name,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NAME | CHARACTER | Returns the program ID |
| NOERR | INTEGER | Returns an error number |
TQGTPI returns the ID of the program. Every program that is able to read ChemApp transparent data-files is identified with a 4-letter mnemonic ID that is retrieved using TQGTPI. In case of a program that is linked against the full version of ChemApp, the program ID would be CAFU, if it is linked against ChemApp light instead, the program ID is CALI.
- See
- TQGTID
2.8 TQGTHI
GET-HASP-INFO
Use TQGTHI to get the HASP dongle type and id.
Added for ChemApp version 4.1.4
Synopsis
- FORTRAN:
- CALL TQGTHI(HASPT,HASPID,NOERR)
- C:
- tqgthi(haspt,&haspid,&noerr);
- Pascal:
- tqgthi(haspt,haspid,noerr);
- Basic:
- Call tqgthi(haspt,haspid,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| HASPT | CHARACTER | Returns the HASP dongle type |
| HASPID | INTEGER | Returns the HASP dongle id |
| NOERR | INTEGER | Returns an error number |
If a version of ChemApp is used that requires a dongle (HASP hardware key), the dongle contains the necessary licensing information. In this case, TQGTHI can be used to retrieve information on the dongle type and the dongle id. This information, together with the information retrieved by TQGTED, TQGTPI, TQGTNM,and TQGTID is necessary if GTT-Technologies is contacted regarding an update of the license.
- See also
- TQGTED
- See
- TQGTID
2.9 TQGTED
GET-EXPIRATION-DATE
Use TQGTED to get the expiration date of the ChemApp license.
Added for ChemApp version 4.1.4
Synopsis
- FORTRAN:
- CALL TQGTED(EDMON,EDYEAR,NOERR)
- C:
- tqgted(&edmon,&edyear,&noerr);
- Pascal:
- tqgted(edmon,edyear,noerr);
- Basic:
- Call tqgted(edmon,edyear,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| EDMON | INTEGER | Returns the month part of the expiration date |
| EDYEAR | INTEGER | Returns the year part of the expiration date |
| NOERR | INTEGER | Returns an error number |
If a version of ChemApp is used that requires a dongle (HASP hardware key), the dongle contains the necessary licensing information. In this case, TQGTED can be used to retrieve the expiration date of the ChemApp license. This information, together with the information retrieved by TQGTHI, TQGTPI, TQGTNM,and TQGTID is necessary if GTT-Technologies is contacted regarding an update of the license.
In case the ChemApp version used does not require a dongle, or if ChemApp is used under a perpetual license, a default date of 12/2025 (December 2025) or later is used.
- See also
- TQGTHI
- See
- TQGTID
2.10 TQCONF
SET-CONFIGURATION-OPTION
Use TQCONF to set a configuration option.
Added for ChemApp version 6.3.4
Synopsis
- FORTRAN:
- CALL TQCONF(OPTION,VALUEA,VALUEB,VALUEC,NOERR)
- C:
- tqconf(option,valuea,valueb,valuec,&noerr);
- Pascal:
- tqconf(option,valuea,valueb,valuec,noerr);
- Basic:
- Call tqconf(option,valuea,valueb,valuec,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Sets configuration option |
| VALUEA | INTEGER | Sets configuration value A |
| VALUEB | INTEGER | Sets configuration value B |
| VALUEC | INTEGER | Sets configuration value C |
| NOERR | INTEGER | Returns an error number |
TQCONF is used to set configuration options, the following options are currently available (for details please see Table TQCONF option):
Semi-automatic charge balance correction (option E)
In iterative calculations, where the input to an equilibrium calculation is based on the output of a previous equilibrium calculation, electroneutrality in phases with phase-internal electrons might not be given. A typical reason for this are rounding errors.
When global conditions are used (see Setting initial conditions), this is not difficult to fix, since the amount of system components, and thus also the amounts of the electron(s), can be directly set with TQSETC. When streams are used, this is not as easily done, for this reason the option of a semi-automatic charge balance correction was included.
Calling TQCONF with the option E instructs ChemApp to set the amount of electrons before every subsequent equilibrium calculation to zero. This can be done either on a per-electron basis by referencing them using their system component index number, or for all electrons by passing a zero for the system component index number (see Table TQCONF option).
Note that the prerequisite for semi-automatic charge balance correction is that the electron(s) in question explicitely appear as system components in the data-file.
Calling TQREMC with a value of -1 or -2 removes any configuration option set previously.
! Using TQCONF to set up semi-automatic charge balance correction PROGRAM CAF34 IMPLICIT NONE INTEGER NOERR, IAQU, IGAS, IPC, IEA DOUBLE PRECISION TPAQU(2), VALS(2) ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNA('water.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Set temperature and pressure units CALL TQCSU('T ', 'C ', NOERR) CALL TQCSU('p ', 'atm ', NOERR) ! Define an input stream 'STREAM_AQUEOUS' at 25 C and 1 atm TPAQU(1) = 25.D0 TPAQU(2) = 1.D0 CALL TQSTTP('STREAM_AQUEOUS', TPAQU, NOERR) ! Define content of this stream: 1 liter (55.508 mole) of water CALL TQINP('aqueous ', IAQU, NOERR) CALL TQINPC('H2O_liquid ', IAQU, IPC, NOERR) CALL TQSTCA('STREAM_AQUEOUS ', IAQU, IPC, 55.508D0, NOERR) ! Set temperature and total pressure to 25 C and 1 atm CALL TQSTEC('P ', 0, 1.D0, NOERR) CALL TQSTEC('T ', 0, 105.D0, NOERR) ! Perform a formation phase target calculation with the gas phase as ! the target phase. Note that the gas phase is always the phase with ! index number 1, irrespective of whether it is called 'GAS', ! 'gas_ideal', or, as in this case, 'gas_real' CALL TQSTEC('A ', 1, 0.D0, NOERR) ! Display what we have set so far CALL TQSHOW(NOERR)
SYSTEM UNITS:
Pressure : atm
Volume : dm3
Temperature : C
Energy : J
Amount : mol
T = 105.00000000 C
P = 1 atm
STREAM NAME STREAM NUMBER TEMPERATURE/C PRESSURE/atm
STREAM_AQUEOUS
1 25.00 1.0000E+00
STREAM NUMBER STREAM CONSTITUENT
1 H2O_liquid/aqueous/
AMOUNT/mol = 5.55080000000000E+01
INACTIVE ELECTRON CONSTRAINT
FORMATION TARGET PHASE: gas_real
EQUILIBRIUM AMOUNT/mol = 0.0000E+00
! In the above output, note the line 'INACTIVE ELECTRON ! CONSTRAINT', i.e. no charge balance correction by default. ! Calculate the equilibrium, specifying temperature as the target ! variable CALL TQCEL('T ', 0, 0, VALS, NOERR)
*T = 100.00 C
P = 1 atm
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/C PRESSURE/atm STREAM
H2O_liquid/aqueous/ 5.5508E+01 25.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: gas_real mol atm
H2O 0.0000E+00 1.0000E+00 9.8754E-01
H2 0.0000E+00 1.1802E-21 1.1950E-21
O2 0.0000E+00 6.1055E-22 6.1591E-22
OH 0.0000E+00 2.0291E-26 2.0038E-26
HOOH 0.0000E+00 3.4804E-29 3.4371E-29
HOO 0.0000E+00 7.4430E-35 7.3502E-35
H 0.0000E+00 4.0975E-39 4.0464E-39
O 0.0000E+00 3.8151E-43 3.7675E-43
O3 0.0000E+00 4.1347E-56 4.1475E-56
TOTAL: 0.0000E+00 1.0000E+00 1.0000E+00
PHASE: aqueous mol MOLALITY ACTIVITY
H2O_liquid 5.5508E+01 5.5508E+01 1.0000E+00
H[+] 7.3345E-07 7.3346E-07 7.3346E-07
H2 1.0812E-24 1.0812E-24 1.0812E-24
O2 5.2886E-25 5.2886E-25 5.2886E-25
OH[-] 7.3345E-07 7.3346E-07 7.3346E-07
HO2[-] 4.4778E-31 4.4778E-31 4.4778E-31
HOOH 2.3471E-26 2.3471E-26 2.3471E-26
TOTAL: 5.5508E+01 1.0000E+00
mol ACTIVITY
H2O_Ice(s) T 0.0000E+00 4.0025E-01
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
3.43097E+01 3.14178E+05 9.39802E+02 -3.27709E+05 0.00000E+00
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
4.21825E+03 -1.55517E+07 4.82259E+03 -1.73512E+07 0.00000E+00
Properties for aqueous:
pH = 6.1346
Eh/V = 0.32014
Total solute molality = 1.4669E-06
Ionic strength = 7.3346E-07
Osmotic coefficient = 1
Mole fraction of system components:
gas_real aqueous
O 0.33333 0.33333
H 0.66667 0.66667
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
! As seen in the previous table, the gas phase over pure water reaches ! a pressure of 1 atm at 100 degrees Celsius. The gas phase amount is ! still zero, as this is the condition for a standard formation phase ! target calculation: the activity (or, in case of a gas phase, the ! fugacity) of the target phase is unity, the phase amount is ! zero. Just like any other phase, a gas phase with an ! activity/fugacity less then unity is not considered stable and thus ! does not have a positive amount. ! Set the equilibrium temperature to 105 degrees Celsius CALL TQSTEC('T ', 0, 105.D0, NOERR) ! Calculate the equilibrium CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 105 C
P = 1 atm
V = 1702.0 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/C PRESSURE/atm STREAM
H2O_liquid/aqueous/ 5.5508E+01 25.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: gas_real mol atm
H2O 5.5508E+01 1.0000E+00 9.8824E-01
H2 1.3219E-19 2.3814E-21 2.4098E-21
O2 6.6093E-20 1.1907E-21 1.2005E-21
OH 2.6353E-24 4.7476E-26 4.6918E-26
HOOH 4.2286E-27 7.6180E-29 7.5284E-29
HOO 1.1498E-32 2.0714E-34 2.0470E-34
H 8.1923E-37 1.4759E-38 1.4585E-38
O 8.5669E-41 1.5434E-42 1.5252E-42
O3 1.1464E-53 2.0654E-55 2.0713E-55
TOTAL: 5.5508E+01 1.0000E+00 1.0000E+00
PHASE: aqueous mol MOLALITY ACTIVITY
H2O_liquid 0.0000E+00 5.5508E+01 8.4106E-01
H[+] 0.0000E+00 8.7350E-07 7.3467E-07
H2 0.0000E+00 2.6604E-24 2.2376E-24
O2 0.0000E+00 1.2340E-24 1.0378E-24
OH[-] 0.0000E+00 8.7350E-07 7.3467E-07
HO2[-] 0.0000E+00 9.2402E-31 7.7716E-31
HOOH 0.0000E+00 4.5929E-26 3.8629E-26
TOTAL: 0.0000E+00 8.4106E-01
mol ACTIVITY
H2O_Ice(s) T 0.0000E+00 3.2254E-01
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
-2.18727E+03 2.58330E+06 7.02064E+03 -3.82181E+05 1.70205E+03
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.99667E+03 -1.32826E+07 1.09034E+04 -1.74057E+07 1.70205E+03
Mole fraction of system components:
gas_real
O 0.33333
H 0.66667
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
! At 5 degrees above the boiling point of water at standard ! conditions, only the gas phase is stable. ! Artificially add an excess amount of OH[-] to the water, this will ! make it impossible for ChemApp to satisfy the electroneutrality ! condition. Note that this is an effect that might occur due to ! rounding errors in iterative calculations. CALL TQINPC('OH[-] ', IAQU, IPC, NOERR) CALL TQSTCA('STREAM_AQUEOUS ', IAQU, IPC, 1.D-7, NOERR) ! Calculate the equilibrium CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 105 C
P = 1 atm
V = 1702.0 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/C PRESSURE/atm STREAM
H2O_liquid/aqueous/ 5.5508E+01 25.00 1.0000E+00 1
OH[-]/aqueous/ 1.0000E-07 25.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: gas_real mol atm
H2O 5.5508E+01 1.0000E+00 9.8824E-01
H2 1.3219E-19 2.3814E-21 2.4098E-21
O2 6.6093E-20 1.1907E-21 1.2005E-21
OH 2.6353E-24 4.7476E-26 4.6918E-26
HOOH 4.2286E-27 7.6180E-29 7.5284E-29
HOO 1.1498E-32 2.0714E-34 2.0470E-34
H 8.1923E-37 1.4759E-38 1.4585E-38
O 8.5669E-41 1.5434E-42 1.5252E-42
O3 1.1464E-53 2.0654E-55 2.0713E-55
TOTAL: 5.5508E+01 1.0000E+00 1.0000E+00
PHASE: aqueous mol MOLALITY ACTIVITY
H2O_liquid 5.7773E-07 5.5508E+01 8.4106E-01
H[+] 5.8467E-22 5.6175E-14 5.6175E-14
H2 2.3289E-32 2.2376E-24 2.2376E-24
O2 1.0802E-32 1.0378E-24 1.0378E-24
OH[-] 1.0000E-07 9.6080E+00 9.6080E+00
HO2[-] 1.0578E-31 1.0164E-23 1.0164E-23
HOOH 4.0205E-34 3.8629E-26 3.8629E-26
TOTAL: 6.7773E-07 1.0000E+00
mol ACTIVITY
H2O_Ice(s) T 0.0000E+00 3.2254E-01
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
-2.18727E+03 2.58330E+06 7.02064E+03 -3.82181E+05 1.70205E+03
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.99667E+03 -1.32826E+07 1.09034E+04 -1.74057E+07 1.70205E+03
Properties for aqueous:
pH = 13.250
Eh/V =-0.22092
Total solute molality = 9.6080
Ionic strength = 4.8040
Osmotic coefficient = 1
Mole fraction of system components:
gas_real aqueous
O 0.33333 0.33333
H 0.66667 0.61748
EA 0 4.9184E-02
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
! The results from the equilibrium calculation now show that liquid ! water is stable, which is definitely incorrect at 105 C. Due to the ! artificially introduced excess amount of OH[-], the Gibbs energy ! minimizer has no choice but to add the water phase to the set of ! stable phases, otherwise the mass balance cannot be satisfied. Note ! also that the excess electrons ('EA') are documented in the table ! "Mole fraction of system components" at the bottom of the output ! above. ! ChemApp can resolve such cases semi-automatically, if it is ! instructed via the subroutine TQCONF to apply a charge balance ! correction. ! OPTION 1: Configure ChemApp to perform semi-automatic charge balance ! correction for one electron spcifically: ! ! Get the system component index number for the aqueous electron ! 'EA' CALL TQINSC('EA ', IEA, NOERR) ! Configure ChemApp to perform semi-automatic charge balance ! correction for this electron CALL TQCONF('E ', IEA, 0, 0, NOERR) ! OPTION 2: Configure ChemApp to perform semi-automatic charge balance ! correction for *all* electrons: ! ! ChemApp can easily be configured to perform the charge balance ! correction for all electrons, in case the data-file contains more ! than one phase-internal electron. CALL TQCONF('E ', 0, 0, 0, NOERR) ! Display what we have set so far CALL TQSHOW(NOERR)
SYSTEM UNITS:
Pressure : atm
Volume : dm3
Temperature : C
Energy : J
Amount : mol
T = 105.00000000 C
P = 1 atm
STREAM NAME STREAM NUMBER TEMPERATURE/C PRESSURE/atm
STREAM_AQUEOUS
1 25.00 1.0000E+00
STREAM NUMBER STREAM CONSTITUENT
1 H2O_liquid/aqueous/
AMOUNT/mol = 5.55080000000000E+01
1 OH[-]/aqueous/
AMOUNT/mol = 1.00000000000000E-07
ACTIVE ELECTRON CONSTRAINT
! In the above output, note that the line 'ACTIVE ELECTRON CONSTRAINT' ! is now present ! Calculate the equilibrium CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 105 C
P = 1 atm
V = 1702.0 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/C PRESSURE/atm STREAM
H2O_liquid/aqueous/ 5.5508E+01 25.00 1.0000E+00 1
OH[-]/aqueous/ 1.0000E-07 25.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: gas_real mol atm
H2O 5.5508E+01 1.0000E+00 9.8824E-01
O2 2.5000E-08 4.5039E-10 4.5409E-10
HOOH 2.6007E-21 4.6853E-23 4.6302E-23
OH 2.0667E-21 3.7232E-23 3.6794E-23
HOO 5.5456E-24 9.9906E-26 9.8731E-26
H2 2.1493E-25 3.8720E-27 3.9182E-27
O 5.2689E-35 9.4921E-37 9.3805E-37
O3 2.6670E-36 4.8048E-38 4.8185E-38
H 1.0446E-39 1.8819E-41 1.8598E-41
TOTAL: 5.5508E+01 1.0000E+00 1.0000E+00
PHASE: aqueous mol MOLALITY ACTIVITY
H2O_liquid 0.0000E+00 5.5508E+01 8.4106E-01
H[+] 0.0000E+00 8.7350E-07 7.3467E-07
H2 0.0000E+00 4.3257E-30 3.6382E-30
O2 0.0000E+00 4.6675E-13 3.9256E-13
OH[-] 0.0000E+00 8.7350E-07 7.3467E-07
HO2[-] 0.0000E+00 5.6830E-25 4.7797E-25
HOOH 0.0000E+00 2.8248E-20 2.3758E-20
TOTAL: 0.0000E+00 8.4106E-01
mol ACTIVITY
H2O_Ice(s) T 0.0000E+00 3.2254E-01
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
-2.18727E+03 2.58330E+06 7.02064E+03 -3.82181E+05 1.70205E+03
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.99667E+03 -1.32826E+07 1.09034E+04 -1.74057E+07 1.70205E+03
Mole fraction of system components:
gas_real
O 0.33333
H 0.66667
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
! Note that liquid water is no longer stable. END
/* Program cac34 */ /* Using TQCONF to set up semi-automatic charge balance correction */ #include "cacint.h" int main() { LI noerr, iAQU, iGAS, iPC, iEA; DB TP_AQU[2], vals[2]; /* Initialise ChemApp */ tqini(&noerr); /* Open data-file for reading */ tqopna("water.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Set temperature and pressure units */ tqcsu("T ", "C ", &noerr); tqcsu("p ", "atm ", &noerr); /* Define an input stream "STREAM_AQUEOUS" at 25 C and 1 atm */ TP_AQU[0] = 25.0; TP_AQU[1] = 1.0; tqsttp("STREAM_AQUEOUS", TP_AQU, &noerr); /* Define content of this stream: 1 liter (55.508 mole) of water */ tqinp("aqueous ", &iAQU, &noerr); tqinpc("H2O_liquid ", iAQU, &iPC, &noerr); tqstca("STREAM_AQUEOUS ", iAQU, iPC, 55.508, &noerr); /* Set temperature and total pressure to 25 C and 1 atm */ tqstec("P ", 0, 1.0, &noerr); tqstec("T ", 0, 105.0, &noerr); /* Perform a formation phase target calculation with the gas phase as the target phase. Note that the gas phase is always the phase with index number 1, irrespective of whether it is called 'GAS', 'gas_ideal', or, as in this case, 'gas_real' */ tqstec("A ", 1, 0.0, &noerr); /* Display what we have set so far */ tqshow(&noerr);
SYSTEM UNITS:
Pressure : atm
Volume : dm3
Temperature : C
Energy : J
Amount : mol
T = 105.00000000 C
P = 1 atm
STREAM NAME STREAM NUMBER TEMPERATURE/C PRESSURE/atm
STREAM_AQUEOUS
1 25.00 1.0000E+00
STREAM NUMBER STREAM CONSTITUENT
1 H2O_liquid/aqueous/
AMOUNT/mol = 5.55080000000000E+01
INACTIVE ELECTRON CONSTRAINT
FORMATION TARGET PHASE: gas_real
EQUILIBRIUM AMOUNT/mol = 0.0000E+00
/* In the above output, note the line 'INACTIVE ELECTRON CONSTRAINT', i.e. no charge balance correction by default. Calculate the equilibrium, specifying temperature as the target variable */ tqcel("T ", 0, 0, vals, &noerr);
*T = 100.00 C
P = 1 atm
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/C PRESSURE/atm STREAM
H2O_liquid/aqueous/ 5.5508E+01 25.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: gas_real mol atm
H2O 0.0000E+00 1.0000E+00 9.8754E-01
H2 0.0000E+00 1.1802E-21 1.1950E-21
O2 0.0000E+00 6.1055E-22 6.1591E-22
OH 0.0000E+00 2.0291E-26 2.0038E-26
HOOH 0.0000E+00 3.4804E-29 3.4371E-29
HOO 0.0000E+00 7.4430E-35 7.3502E-35
H 0.0000E+00 4.0975E-39 4.0464E-39
O 0.0000E+00 3.8151E-43 3.7675E-43
O3 0.0000E+00 4.1347E-56 4.1475E-56
TOTAL: 0.0000E+00 1.0000E+00 1.0000E+00
PHASE: aqueous mol MOLALITY ACTIVITY
H2O_liquid 5.5508E+01 5.5508E+01 1.0000E+00
H[+] 7.3345E-07 7.3346E-07 7.3346E-07
H2 1.0812E-24 1.0812E-24 1.0812E-24
O2 5.2886E-25 5.2886E-25 5.2886E-25
OH[-] 7.3345E-07 7.3346E-07 7.3346E-07
HO2[-] 4.4778E-31 4.4778E-31 4.4778E-31
HOOH 2.3471E-26 2.3471E-26 2.3471E-26
TOTAL: 5.5508E+01 1.0000E+00
mol ACTIVITY
H2O_Ice(s) T 0.0000E+00 4.0025E-01
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
3.43097E+01 3.14178E+05 9.39802E+02 -3.27709E+05 0.00000E+00
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
4.21825E+03 -1.55517E+07 4.82259E+03 -1.73512E+07 0.00000E+00
Properties for aqueous:
pH = 6.1346
Eh/V = 0.32014
Total solute molality = 1.4669E-06
Ionic strength = 7.3346E-07
Osmotic coefficient = 1
Mole fraction of system components:
gas_real aqueous
O 0.33333 0.33333
H 0.66667 0.66667
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
/* As seen in the previous table, the gas phase over pure water reaches a pressure of 1 atm at 100 degrees Celsius. The gas phase amount is still zero, as this is the condition for a standard formation phase target calculation: the activity (or, in case of a gas phase, the fugacity) of the target phase is unity, the phase amount is zero. Just like any other phase, a gas phase with an activity/fugacity less then unity is not considered stable and thus does not have a positive amount. */ /* Set the equilibrium temperature to 105 degrees Celsius */ tqstec("T ", 0, 105.0, &noerr); /* Calculate the equilibrium */ tqcel(" ", 0, 0, vals, &noerr);
T = 105 C
P = 1 atm
V = 1702.0 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/C PRESSURE/atm STREAM
H2O_liquid/aqueous/ 5.5508E+01 25.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: gas_real mol atm
H2O 5.5508E+01 1.0000E+00 9.8824E-01
H2 1.3219E-19 2.3814E-21 2.4098E-21
O2 6.6093E-20 1.1907E-21 1.2005E-21
OH 2.6353E-24 4.7476E-26 4.6918E-26
HOOH 4.2286E-27 7.6180E-29 7.5284E-29
HOO 1.1498E-32 2.0714E-34 2.0470E-34
H 8.1923E-37 1.4759E-38 1.4585E-38
O 8.5669E-41 1.5434E-42 1.5252E-42
O3 1.1464E-53 2.0654E-55 2.0713E-55
TOTAL: 5.5508E+01 1.0000E+00 1.0000E+00
PHASE: aqueous mol MOLALITY ACTIVITY
H2O_liquid 0.0000E+00 5.5508E+01 8.4106E-01
H[+] 0.0000E+00 8.7350E-07 7.3467E-07
H2 0.0000E+00 2.6604E-24 2.2376E-24
O2 0.0000E+00 1.2340E-24 1.0378E-24
OH[-] 0.0000E+00 8.7350E-07 7.3467E-07
HO2[-] 0.0000E+00 9.2402E-31 7.7716E-31
HOOH 0.0000E+00 4.5929E-26 3.8629E-26
TOTAL: 0.0000E+00 8.4106E-01
mol ACTIVITY
H2O_Ice(s) T 0.0000E+00 3.2254E-01
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
-2.18727E+03 2.58330E+06 7.02064E+03 -3.82181E+05 1.70205E+03
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.99667E+03 -1.32826E+07 1.09034E+04 -1.74057E+07 1.70205E+03
Mole fraction of system components:
gas_real
O 0.33333
H 0.66667
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
/* At 5 degrees above the boiling point of water at standard conditions, only the gas phase is stable. */ /* Artificially add an excess amount of OH[-] to the water, this will make it impossible for ChemApp to satisfy the electroneutrality condition. Note that this is an effect that might occur due to rounding errors in iterative calculations. */ tqinpc("OH[-] ", iAQU, &iPC, &noerr); tqstca("STREAM_AQUEOUS ", iAQU, iPC, 1.0E-7, &noerr); /* Calculate the equilibrium */ tqcel(" ", 0, 0, vals, &noerr);
T = 105 C
P = 1 atm
V = 1702.0 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/C PRESSURE/atm STREAM
H2O_liquid/aqueous/ 5.5508E+01 25.00 1.0000E+00 1
OH[-]/aqueous/ 1.0000E-07 25.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: gas_real mol atm
H2O 5.5508E+01 1.0000E+00 9.8824E-01
H2 1.3219E-19 2.3814E-21 2.4098E-21
O2 6.6093E-20 1.1907E-21 1.2005E-21
OH 2.6353E-24 4.7476E-26 4.6918E-26
HOOH 4.2286E-27 7.6180E-29 7.5284E-29
HOO 1.1498E-32 2.0714E-34 2.0470E-34
H 8.1923E-37 1.4759E-38 1.4585E-38
O 8.5669E-41 1.5434E-42 1.5252E-42
O3 1.1464E-53 2.0654E-55 2.0713E-55
TOTAL: 5.5508E+01 1.0000E+00 1.0000E+00
PHASE: aqueous mol MOLALITY ACTIVITY
H2O_liquid 5.7773E-07 5.5508E+01 8.4106E-01
H[+] 5.8467E-22 5.6175E-14 5.6175E-14
H2 2.3289E-32 2.2376E-24 2.2376E-24
O2 1.0802E-32 1.0378E-24 1.0378E-24
OH[-] 1.0000E-07 9.6080E+00 9.6080E+00
HO2[-] 1.0578E-31 1.0164E-23 1.0164E-23
HOOH 4.0205E-34 3.8629E-26 3.8629E-26
TOTAL: 6.7773E-07 1.0000E+00
mol ACTIVITY
H2O_Ice(s) T 0.0000E+00 3.2254E-01
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
-2.18727E+03 2.58330E+06 7.02064E+03 -3.82181E+05 1.70205E+03
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.99667E+03 -1.32826E+07 1.09034E+04 -1.74057E+07 1.70205E+03
Properties for aqueous:
pH = 13.250
Eh/V =-0.22092
Total solute molality = 9.6080
Ionic strength = 4.8040
Osmotic coefficient = 1
Mole fraction of system components:
gas_real aqueous
O 0.33333 0.33333
H 0.66667 0.61748
EA 0 4.9184E-02
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
/* The results from the equilibrium calculation now show that liquid water is stable, which is definitely incorrect at 105 C. Due to the artificially introduced excess amount of OH[-], the Gibbs energy minimizer has no choice but to add the water phase to the set of stable phases, otherwise the mass balance cannot be satisfied. Note also that the excess electrons ('EA') are documented in the table "Mole fraction of system components" at the bottom of the output above. */ /* ChemApp can resolve such cases semi-automatically, if it is instructed via the subroutine TQCONF to apply a charge balance correction. */ /* OPTION 1: Configure ChemApp to perform semi-automatic charge balance correction for one electron spcifically: Get the system component index number for the aqueous electron 'EA' */ tqinsc("EA ", &iEA, &noerr); /* Configure ChemApp to perform semi-automatic charge balance correction for this electron */ tqconf("E ", iEA, 0, 0, &noerr); /* OPTION 2: Configure ChemApp to perform semi-automatic charge balance correction for *all* electrons: ChemApp can easily be configured to perform the charge balance correction for all electrons, in case the data-file contains more than one phase-internal electron. */ tqconf("E ", 0, 0, 0, &noerr); /* Display what we have set so far */ tqshow(&noerr);
SYSTEM UNITS:
Pressure : atm
Volume : dm3
Temperature : C
Energy : J
Amount : mol
T = 105.00000000 C
P = 1 atm
STREAM NAME STREAM NUMBER TEMPERATURE/C PRESSURE/atm
STREAM_AQUEOUS
1 25.00 1.0000E+00
STREAM NUMBER STREAM CONSTITUENT
1 H2O_liquid/aqueous/
AMOUNT/mol = 5.55080000000000E+01
1 OH[-]/aqueous/
AMOUNT/mol = 1.00000000000000E-07
ACTIVE ELECTRON CONSTRAINT
/* In the above output, note that the line 'ACTIVE ELECTRON CONSTRAINT' is now present */ /* Calculate the equilibrium */ tqcel(" ", 0, 0, vals, &noerr);
T = 105 C
P = 1 atm
V = 1702.0 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/C PRESSURE/atm STREAM
H2O_liquid/aqueous/ 5.5508E+01 25.00 1.0000E+00 1
OH[-]/aqueous/ 1.0000E-07 25.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: gas_real mol atm
H2O 5.5508E+01 1.0000E+00 9.8824E-01
O2 2.5000E-08 4.5039E-10 4.5409E-10
HOOH 2.6007E-21 4.6853E-23 4.6302E-23
OH 2.0667E-21 3.7232E-23 3.6794E-23
HOO 5.5456E-24 9.9906E-26 9.8731E-26
H2 2.1493E-25 3.8720E-27 3.9182E-27
O 5.2689E-35 9.4921E-37 9.3805E-37
O3 2.6670E-36 4.8048E-38 4.8185E-38
H 1.0446E-39 1.8819E-41 1.8598E-41
TOTAL: 5.5508E+01 1.0000E+00 1.0000E+00
PHASE: aqueous mol MOLALITY ACTIVITY
H2O_liquid 0.0000E+00 5.5508E+01 8.4106E-01
H[+] 0.0000E+00 8.7350E-07 7.3467E-07
H2 0.0000E+00 4.3257E-30 3.6382E-30
O2 0.0000E+00 4.6675E-13 3.9256E-13
OH[-] 0.0000E+00 8.7350E-07 7.3467E-07
HO2[-] 0.0000E+00 5.6830E-25 4.7797E-25
HOOH 0.0000E+00 2.8248E-20 2.3758E-20
TOTAL: 0.0000E+00 8.4106E-01
mol ACTIVITY
H2O_Ice(s) T 0.0000E+00 3.2254E-01
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
-2.18727E+03 2.58330E+06 7.02064E+03 -3.82181E+05 1.70205E+03
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.99667E+03 -1.32826E+07 1.09034E+04 -1.74057E+07 1.70205E+03
Mole fraction of system components:
gas_real
O 0.33333
H 0.66667
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
/* Note that liquid water is no longer stable. */ return 0; }
| Option | Meaning | Comment |
|---|---|---|
| E | Semi-automatic charge balance correction | In order to have ChemApp set the amount of a specific electron to zero before every equilibrium calculation, set VALUEA to the electron's system component index number. In order to set the amount of all electrons to zero, set VALUEA to zero. This will affect all system components with names starting with 'e(', 'E(', or 'EA' |
2.11 TQSIZE
GET-ARRAY-SIZES
Use TQSIZE to get the internal array dimensions of ChemApp.
Added for ChemApp version 1.1.3
Synopsis
- FORTRAN:
- CALL TQSIZE(NA,NB,NC,ND,NE,NF,NG,NH,NI,NJ,NK,NOERR)
- C:
- tqsize(&na,&nb,&nc,&nd,&ne,&nf,&ng,&nh,&ni,&nj,&nk,&noerr);
- Pascal:
- tqsize(na,nb,nc,nd,ne,nf,ng,nh,ni,nj,nk,noerr);
- Basic:
- Call tqsize(na,nb,nc,nd,ne,nf,ng,nh,ni,nj,nk,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NA | INTEGER | Returns the maximum number of constituents |
| NB | INTEGER | Returns the maximum number of system components |
| NC | INTEGER | Returns the maximum number of mixture phases |
| ND | INTEGER | Returns the maximum number of excess Gibbs energy coefficients for a mixture phase |
| NE | INTEGER | Returns the maximum number of excess magnetic coefficients for a mixture phase |
| NF | INTEGER | Returns the maximum number of sublattices for a mixture phase |
| NG | INTEGER | Returns the maximum number of constituents of a sublattice |
| NH | INTEGER | Returns the maximum number of oxide constituents of a phase described by the Gaye-Kapoor-Frohberg or modified quasichemical formalisms |
| NI | INTEGER | Returns the maximum number of Gibbs energy/heat capacity equations for a constituent |
| NJ | INTEGER | Returns the maximum number of Gibbs energy/heat capacity equations |
| NK | INTEGER | Returns the maximum number of constituents with P,T-dependent molar volumes |
| NOERR | INTEGER | Returns an error number |
TQSIZE returns the dimensions of a number of internal arrays. Among other purposes, it can be used to verify whether one is working with the standard or extended version of ChemApp.
- See also
- TQUSED
! Display the sizes of selected ChemApp internal arrays PROGRAM CAF4 IMPLICIT NONE INTEGER NOERR INTEGER LA,LB,LC,LD,LE,LF,LG,LH,LI,LJ,LK ! Initialise ChemApp CALL TQINI(NOERR) ! Get array sizes CALL TQSIZE(LA,LB,LC,LD,LE,LF,LG,LH,LI,LJ,LK,NOERR) 50 FORMAT(1X,A,I3) WRITE(UNIT=*,FMT=50) 'Maximum number of constituents: ',LA WRITE(UNIT=*,FMT=50) 'Maximum number of system components: ',LB WRITE(UNIT=*,FMT=50) 'Maximum number of mixture phases: ',LC WRITE(UNIT=*,FMT=50) 'Maximum number of sublattices for a '// * 'mixture phase: ',LF END
Maximum number of constituents: *** Maximum number of system components: 48 Maximum number of mixture phases: 44 Maximum number of sublattices for a mixture phase: 5
/* Program cac4 */ /* Display the sizes of selected ChemApp internal arrays */ #include "cacint.h" int main() { LI noerr, la,lb,lc,ld,le,lf,lg,lh,li,lj,lk; /* Initialise ChemApp */ tqini(&noerr); /* Get array sizes */ tqsize(&la,&lb,&lc,&ld,&le,&lf,&lg,&lh,&li,&lj,&lk,&noerr); printf("Internal array sizes of this version of ChemApp:\n" "Maximum number of constituents: %li\n" "Maximum number of system components: %li\n" "Maximum number of mixture phases: %li\n" "Maximum number of sublattices for a mixture phase: %li\n", la,lb,lc,lf); return 0; }
Internal array sizes of this version of ChemApp: Maximum number of constituents: 1600 Maximum number of system components: 48 Maximum number of mixture phases: 44 Maximum number of sublattices for a mixture phase: 5
2.12 TQUSED
GET-CURRENTLY-USED-DIMENSIONS
Use TQUSED to get the dimensions of the currently loaded thermochemical system.
Added for ChemApp version 4.0.0
Synopsis
- FORTRAN:
- CALL TQUSED(NA,NB,NC,ND,NE,NF,NG,NH,NI,NJ,NK,NOERR)
- C:
- tqused(&na,&nb,&nc,&nd,&ne,&nf,&ng,&nh,&ni,&nj,&nk,&noerr);
- Pascal:
- tqused(na,nb,nc,nd,ne,nf,ng,nh,ni,nj,nk,noerr);
- Basic:
- Call tqused(na,nb,nc,nd,ne,nf,ng,nh,ni,nj,nk,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NA | INTEGER | Returns the maximum number of constituents |
| NB | INTEGER | Returns the maximum number of system components |
| NC | INTEGER | Returns the maximum number of mixture phases |
| ND | INTEGER | Returns the maximum number of excess Gibbs energy coefficients for a mixture phase |
| NE | INTEGER | Returns the maximum number of excess magnetic coefficients for a mixture phase |
| NF | INTEGER | Returns the maximum number of sublattices for a mixture phase |
| NG | INTEGER | Returns the maximum number of constituents of a sublattice |
| NH | INTEGER | Returns the maximum number of oxide constituents of a phase described by the Gaye-Kapoor-Frohberg or modified quasichemical formalisms |
| NI | INTEGER | Returns the maximum number of Gibbs energy/heat capacity equations for a constituent |
| NJ | INTEGER | Returns the maximum number of Gibbs energy/heat capacity equations |
| NK | INTEGER | Returns the maximum number of constituents with P,T-dependent molar volumes |
| NOERR | INTEGER | Returns an error number |
TQUSED resembles TQSIZE and mirrors its parameters. The difference is that TQUSED returns the values for the currently loaded thermochemical system, whereas TQSIZE returns the maximum values for these parameters, and thus indicates the size of the internal arrays in ChemApp.
TQUSED was introduced as a support subroutine for transparent data-files, but might be used by a ChemApp programmer to check how much larger a thermochemical system contained in a data-file can be before the internal limits of the ChemApp library used (light, standard, or extended) are reached. In this case the parameters NA through NK returned by TQUSED would be compared to the parameters of the same name TQSIZE returns.
- See also
- TQSIZE
- See
- TQSIZE
2.13 TQGIO
GET-VALUE-OF-INPUT-OUTPUT-OPTION
Use TQGIO to get the unit number and language used for error messages, and the unit number for the thermodynamic data-file and optional output lists, respectively.
Synopsis
- FORTRAN:
- CALL TQGIO(OPTION,IVAL,NOERR)
- C:
- tqgio(option,&ival,&noerr);
- Pascal:
- tqgio(option,ival,noerr);
- Basic:
- Call tqgio(option,ival,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table Legal input/output options used by TQGIO and TQCIO |
| IVAL | INTEGER | Returns the current unit number or the index for the language used |
| NOERR | INTEGER | Returns an error number |
OPTION is character input identifying the input/output option. Legal values for OPTION are given in Table Legal input/output options used by TQGIO and TQCIO. IVAL is output and equal to the current unit number or to the index for the language used for error messages (see Table Legal input/output options used by TQGIO and TQCIO).
- See also
- TQCIO
! Get values for input/output options PROGRAM CAF5 IMPLICIT NONE INTEGER NOERR, NVALUE ! Initialise ChemApp CALL TQINI(NOERR) ! Get values for input/output options ! Determine which FORTRAN unit is used by TQRFIL for reading the ! thermochemical data-file CALL TQGIO('FILE ', NVALUE, NOERR) 50 FORMAT(1X,A,I3) WRITE(UNIT=*,FMT=50) 'The thermochemical data will be read '// * 'from the file associated with unit ',NVALUE END
The thermochemical data will be read from the file associated with unit 10
/* Program cac5 */ /* Get values for input/output options */ #include "cacint.h" int main() { LI noerr, iovalue; /* Initialise ChemApp */ tqini(&noerr); /* Get values for input/output options Determine which FORTRAN unit is used by tqrfil for reading the thermochemical data-file */ tqgio("FILE", &iovalue, &noerr); printf("The thermochemical data will be read from the file " "associated with unit %li\n", iovalue); return 0; }
The thermochemical data will be read from the file associated with unit 10
| Option | Meaning | Default value |
|---|---|---|
| FILE | Unit from which the thermodynamic data-file is read | 10 |
| ERROR | Unit to which error messages are directed | 6 |
| LIST | Unit to which optional equilibrium output lists are directed | 6 |
| LANGUAGE | Language used for error messages (1=English, 2=German, 3=French, 4=Swedish, 5=Finnish) | 1 |
2.14 TQCIO
CHANGE-VALUE-OF-INPUT-OUTPUT-OPTION
Using TQCIO, input and output from ChemApp can be redirected to another unit than the default, or a different language be selected for the error messages than the default (see Table Legal input/output options used by TQGIO and TQCIO ).
Synopsis
- FORTRAN:
- CALL TQCIO(OPTION,IVAL,NOERR)
- C:
- tqcio(option,ival,&noerr);
- Pascal:
- tqcio(option,ival,noerr);
- Basic:
- Call tqcio(option,ival,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table Legal input/output options used by TQGIO and TQCIO |
| IVAL | INTEGER | Set to a legal unit number, or to a language index |
| NOERR | INTEGER | Returns an error number |
OPTION and IVAL are both input, and are used to identify the input/output option (see Table Legal input/output options used by TQGIO and TQCIO). Unit numbers entered must be <= 10 or >= 20 to be valid. Error messages will not be output if IVAL is set to zero for option ERROR.
A file has to be opened under this new unit number in the application program if default values are changed for the options ERROR and LIST. TQOPEN and TQCLOS should be used for this purpose.
- See also
- TQGIO
! Change values of input/output options PROGRAM CAF6 IMPLICIT NONE INTEGER NOERR, NPHASE ! Initialise ChemApp CALL TQINI(NOERR) ! Change the language for error messages to Swedish CALL TQCIO('LANGUAGE ', 4, NOERR) ! Trigger a ChemApp error by trying to get the number of phases ! before even having read a thermochemical data-file CALL TQNOP(NPHASE, NOERR) END
Fel nummer: 104 En fil med termodynamiska data maste foerst inlaesas Felet upptraedde sedan "TQNOP " anropats
/* Program cac6 */ /* Change values of input/output options */ #include "cacint.h" int main() { LI noerr, nphase; /* Initialise ChemApp */ tqini(&noerr); /* Change the language for error messages to Swedish */ tqcio("LANGUAGE", 4, &noerr); /* Trigger a ChemApp error by trying to get the number of phases before even having read a thermochemical data-file */ tqnop(&nphase, &noerr); return 0; }
Fel nummer: 104 En fil med termodynamiska data maste foerst inlaesas Felet upptraedde sedan "TQNOP " anropats
2.15 TQRFIL
READ-DATA-FILE
Reads a thermodynamic data-file in ASCII format.
Synopsis
- FORTRAN:
- CALL TQRFIL(NOERR)
- C:
- tqrfil(&noerr)
- Pascal:
- tqrfil(noerr)
- Basic:
- Call tqrfil(noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NOERR | INTEGER | Returns an error number |
The thermodynamic data-file in ASCII format to be read must be opened in the application program using default unit 10 (see Table Legal input/output options used by TQGIO and TQCIO), unless this has been changed using TQCIO.
For non-FORTRAN programs, TQOPNA and TQCLOS have to be used for opening and closing the data-file.
If the data-file to load is in binary format, use TQRBIN instead; if it is in transparent format, use TQRCST.
- See also
- TQOPNA, TQCLOS, Thermodynamic data
! Read a thermochemical data-file in ASCII format PROGRAM CAF7 IMPLICIT NONE INTEGER NOERR ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) END
/* Program cac7 */ /* Read a thermochemical data-file in ASCII format*/ #include "cacint.h" int main() { LI noerr; /* Initialise ChemApp */ tqini(&noerr); /* Note that tqopna is used to open the thermochemical data-file, instead of a standard C library routine like fopen. The reason is that it is necessary to associate the data-file with a particular FORTRAN unit number (10 by default, see tqgio) so that the FORTRAN subroutine tqrfil can read from the correct file */ tqopna("cosi.dat",10,&noerr); if (noerr) { printf("ERROR: Cannot open data-file."); exit(noerr); } /* Read data-file */ tqrfil(&noerr); /* Close data-file */ /* Again, the routine for closing the data-file is not a standard C library routine like fclose, but a special one to make sure the file that was previously opened under the unit number specified is closed. */ tqclos(10,&noerr); return 0; }
2.16 TQRBIN
READ-BINARY-DATA-FILE
Reads a thermodynamic data-file in binary format.
Added for ChemApp version 2.1.4
Synopsis
- FORTRAN:
- CALL TQRBIN(NOERR)
- C:
- tqrbin(&noerr);
- Pascal:
- tqrbin(noerr);
- Basic:
- Call tqrbin(noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NOERR | INTEGER | Returns an error number |
TQRBIN must be used to read a thermodynamic data-file in binary format. Binary data-files are usually either supplied by GTT-Technologies directly, or created using other software programs (e.g. FACT-Win).
The thermodynamic data-file to be read must be opened in the application program using default unit 10 (see Table Legal input/output options used by TQGIO and TQCIO), unless this has been changed using TQCIO.
For non-FORTRAN programs, TQOPNB and TQCLOS have to be used for opening and closing the data-file.
If the data-file to load is in ASCII (i.e. plain text) format, use TQRFIL instead; if it is in transparent format, use TQRCST.
- See also
- TQOPNB, TQCLOS, Thermodynamic data
! Read a thermochemical data-file in BINARY format ! NOTE: This example program requires a data-file in binary format ! to run, which is not part of a regular distribution of ChemApp ! or ChemApp "light". PROGRAM CAF29 IMPLICIT NONE INTEGER NOERR ! Initialise ChemApp CALL TQINI(NOERR) ! Open binary data-file for reading. CALL TQOPNB('cosi.bin', 10, NOERR) ! Read data-file CALL TQRBIN(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) END
/* Program cac29 */ /* Read a thermochemical data-file in BINARY format*/ /* NOTE: This example program requires a data-file in binary format to run, which is not part of a regular distribution of ChemApp or ChemApp "light". */ #include "cacint.h" int main() { LI noerr; /* Initialise ChemApp */ tqini(&noerr); /* Note that tqopnb is used to open the thermochemical data-file, instead of a standard C library routine like fopen. The reason is that it is necessary to associate the data-file with a particular FORTRAN unit number (10 by default, see tqgio) so that the FORTRAN subroutine tqrbin can read from the correct file */ tqopnb("cosi.bin",10,&noerr); /* Read data-file */ tqrbin(&noerr); /* Close data-file */ /* Again, the routine for closing the data-file is not a standard C library routine like fclose, but a special one to make sure the file that was previously opened under the unit number specified is closed. */ tqclos(10,&noerr); return 0; }
2.17 TQRCST
READ-TRANSPARENT-DATA-FILE
Reads a thermodynamic data-file in transparent format.
Added for ChemApp version 4.0.0
Synopsis
- FORTRAN:
- CALL TQRCST(NOERR)
- C:
- tqrcst(&noerr);
- Pascal:
- tqrcst(noerr);
- Basic:
- Call tqrcst(noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NOERR | INTEGER | Returns an error number |
TQRCST must be used to read a thermodynamic data-file in transparent format. Transparent data-files are usually either supplied by GTT-Technologies directly, or created using other software programs (e.g. FactSage).
The thermodynamic data-file to be read must be opened in the application program using default unit 10 (see Table Legal input/output options used by TQGIO and TQCIO), unless this has been changed using TQCIO.
For both FORTRAN and non-FORTRAN programs, TQOPNT and TQCLOS have to be used for opening and closing the data-file.
If the data-file to load is in ASCII (i.e. plain text) format, use TQRFIL instead; if it is in binary format, use TQRBIN.
- See also
- TQOPNT, TQCLOS, Thermodynamic data, TQGTRH
! Read a thermochemical data-file in TRANSPARENT format, then ! retrieves information on the transparent file header. ! NOTE: This example program requires a data-file in transparent format ! to run, which is not part of a regular distribution of ChemApp ! or ChemApp "light". ! The support function STRLEN returns the total number of characters in ! a string, not counting trailing blanks INTEGER FUNCTION STRLEN(INSTR) CHARACTER INSTR*(*) STRLEN = LEN(INSTR) DO WHILE(INSTR(STRLEN:STRLEN) .EQ. ' ') STRLEN = STRLEN - 1 IF (STRLEN .EQ. 1) RETURN ENDDO RETURN END PROGRAM CAF31 IMPLICIT NONE INTEGER NOERR, STRLEN ! Variables to store information from various transparent file ! header (TFH) fields CHARACTER TFHID*255, TFHUSR*80,TFHNWP*40,TFHNRP*40,TFHREM*80 INTEGER TFHVER, TFHVNW(3), TFHVNR(3), TFHDTC(6), TFHDTE(6) 10 FORMAT(1X,A,I4.4,5(A1,I2.2)) ! Initialise ChemApp CALL TQINI(NOERR) ! Open transparent data-file for reading. CALL TQOPNT('cosiex.cst', 10, NOERR) ! Read data-file CALL TQRCST(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Once the transparent data-file has been read, information on its ! header can be retrieved. CALL TQGTRH(TFHVER, TFHNWP, TFHVNW, TFHNRP, TFHVNR, TFHDTC, * TFHDTE, TFHID, TFHUSR, TFHREM, NOERR) WRITE(UNIT=*, FMT=*) 'Version number of the transparent file ' // * 'header format:', TFHVER WRITE(UNIT=*, FMT=*) 'Name of the program which wrote the ' // * 'data-file: ', TFHNWP(1:STRLEN(TFHNWP)) WRITE(UNIT=*, FMT=*) 'Version number of the writing program: ', * TFHVNW(1), '.', TFHVNW(2), '.', TFHVNW(3) WRITE(UNIT=*, FMT=*) 'Programs which are permitted to read ' // * 'the data-file: ', TFHNRP(1:STRLEN(TFHNRP)) WRITE(UNIT=*, FMT=*) 'Min. version number of the reading ' // * 'program: ', TFHVNR(1), '.', TFHVNR(2), '.', TFHVNR(3) WRITE(UNIT=*, FMT=10) 'File was created on ', * TFHDTC(1),'/', TFHDTC(2),'/',TFHDTC(3),' ', * TFHDTC(4),':',TFHDTC(5),':',TFHDTC(6) WRITE(UNIT=*, FMT=10) 'File will expire on ', * TFHDTE(1),'/', TFHDTE(2),'/',TFHDTE(3),' ', * TFHDTE(4),':',TFHDTE(5),':',TFHDTE(6) WRITE(UNIT=*, FMT=*) 'Licensee''s user ID(s): ', * TFHID(1:STRLEN(TFHID)) WRITE(UNIT=*, FMT=*) 'Licensee''s name: ', * TFHUSR(1:STRLEN(TFHUSR)) WRITE(UNIT=*, FMT=*) 'Remarks: ', * TFHREM(1:STRLEN(TFHREM)) END
Version number of the transparent file header format: 1 Name of the program which wrote the data-file: FactSage Version number of the writing program: 5 . 0 . 0 Programs which are permitted to read the data-file: CAFU,CALI Min. version number of the reading program: -1 . -1 . -1 File was created on 2015/09/09 15:13:52 File will expire on 2036/12/31 12:00:00 Licensee's user ID(s): ???? Licensee's name: GTT - Technologies Remarks: For use with ChemApp example programs
/* Program cac31 */
/* Read a thermochemical data-file in TRANSPARENT format, then
retrieves information on the transparent file header. */
/* NOTE: This example program requires a data-file in transparent format
to run, which is not part of a regular distribution of ChemApp
or ChemApp "light". */
#include "cacint.h"
int main()
{
LI noerr;
/* Variables to store information from various transparent file
header (TFH) fields */
char tfhid[255], tfhusr[80],tfhnwp[40],tfhnrp[40],tfhrem[80];
LI tfhver, tfhvnw[3], tfhvnr[3], tfhdtc[6], tfhdte[6];
/* Initialise ChemApp */
tqini(&noerr);
/* Note that tqopnt is used to open the thermochemical data-file,
instead of a standard C library routine like fopen. The reason
is that it is necessary to associate the data-file with a
particular FORTRAN unit number (10 by default, see tqgio) so
that the FORTRAN subroutine tqrcst can read from the correct
file */
tqopnt("cosiex.cst",10,&noerr);
/* Read data-file */
tqrcst(&noerr);
/* Close data-file */
/* Again, the routine for closing the data-file is not a standard
C library routine like fclose, but a special one to make sure the
file that was previously opened under the unit number specified
is closed. */
tqclos(10,&noerr);
/* Once the transparent data-file has been read, information on its
header can be retrieved.*/
tqgtrh(&tfhver, tfhnwp, tfhvnw, tfhnrp, tfhvnr, tfhdtc, tfhdte,
tfhid, tfhusr, tfhrem, &noerr);
printf("Version number of the transparent file header format: %li\n",
tfhver);
printf("Name of the program which wrote the data-file: %s\n", tfhnwp);
printf("Version number of the writing program: %li.%li.%li\n",
tfhvnw[0],tfhvnw[1],tfhvnw[2]);
printf("Programs which are permitted to read the data-file: %s\n", tfhnrp);
printf("Min. version number of the reading program: %li.%li.%li\n",
tfhvnr[0],tfhvnr[1],tfhvnr[2]);
printf("File was created on %li/%02li/%02li %02li:%02li:%02li \n",
tfhdtc[0],tfhdtc[1],tfhdtc[2],tfhdtc[3],tfhdtc[4],tfhdtc[5]);
printf("File will expire on %li/%02li/%02li %02li:%02li:%02li \n",
tfhdte[0],tfhdte[1],tfhdte[2],tfhdte[3],tfhdte[4],tfhdte[5]);
printf("Licensee's user ID(s): %s\n", tfhid);
printf("Licensee's name: %s\n", tfhusr);
printf("Remarks : %s\n", tfhrem);
return 0;
}
Version number of the transparent file header format: 1 Name of the program which wrote the data-file: FactSage Version number of the writing program: 5.0.0 Programs which are permitted to read the data-file: CAFU,CALI Min. version number of the reading program: 4294967295.4294967295.4294967295 File was created on 2015/09/09 15:13:140720308486196 File will expire on 2036/12/31 12:00:140720308486144 Licensee's user ID(s): ???? Licensee's name: GTT - Technologies Remarks : For use with ChemApp example programs
2.18 TQOPEN
OPEN-FILE
Use TQOPEN to open a file for reading or writing by ChemApp.
TQOPEN has been partially superceded by TQOPNA and TQOPNB in ChemApp versions V3.3.0 and later, see the note below.
Synopsis
- FORTRAN:
- CALL TQOPEN(NAME,UNIT,NOERR)
- C:
- tqopen(name,unit,&noerr);
- Pascal:
- tqopen(name,unit,noerr);
- Basic:
- Call tqopen(name,unit,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NAME | CHARACTER | Set to the filename of the data-file to open |
| UNIT | CHARACTER | Set to the unit number under which it should be opened |
| NOERR | INTEGER | Returns an error number |
This routine is used to open files under a specific FORTRAN unit number. Starting with ChemApp V3.3.0, it should only be used to open files for writing. FORTRAN programmers may use the standard OPEN statement instead, if ChemApp is used as a static library.
There are three standard cases where ChemApp application programs need to open files using TQOPEN:
- TQRFIL and TQRBIN, which read a thermochemical data-file, expect this file to be opened under a specific unit number (10 by default, see Table Legal input/output options used by TQGIO and TQCIO). If you are using ChemApp V3.3.0 or later, you should use TQOPNA and TQOPNB instead to open ASCII and binary thermochemical data-files.
- TQCEL, TQMAPL, and TQSHOW write by default to standard output. If the output from these subroutines should be written to a file instead, the file has to be opened using TQOPEN with the proper unit number (see TQCIO and Table Legal input/output options used by TQGIO and TQCIO).
- Error messages from ChemApp are by default written to standard output as the errors occur. If this is not desired (for instance when the application program has a graphical user interface), the error messages can be redirected to a file, and/or retrieved individually using TQERR. The file to which the error messages would be written in such a case needs to be opened using TQOPEN.
Note that TQOPEN does not check whether the file to be opened already exists or not, since TQOPEN can be used both for opening existing files for reading, and new files for writing. Whether the file to be opened already exists should be checked prior to a call to TQOPEN.
If you receive an Error code 102 or Error code 103 when reading the data-file with TQRFIL, and you find a file of zero bytes length under the name you passed to TQOPEN, this means that this file has just been created with the call to TQOPEN. It didn't exist before, and didn't get shortened, but created instead. It usually means that either the wrong filename was passed to TQOPEN, or the file was not located in the directory where the program expected it. The reason for this behaviour is that TQOPEN can both be used to open existing files for reading, as well as new files for writing.
Both problems mentioned above are resolved if TQOPNA is used instead of TQOPEN to open thermochemical data-files in ASCII format, TQOPNB to open binary data-files, and TQOPNT to open transparent data-files.
A file that has been opened using TQOPEN should subsequently be closed only with TQCLOS.
- See
- TQWSTR
2.19 TQWSTR
WRITE-STRING
Use TQWSTR to write a character string to the Fortran units associated with 'LIST' and 'ERROR'.
Synopsis
- FORTRAN:
- CALL TQWSTR(OPTION,TEXT,NOERR)
- C:
- tqwstr(option,text,&noerr);
- Pascal:
- tqwstr(option,text,noerr);
- Basic:
- Call tqwstr(option,text,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to either 'LIST' or 'ERROR' |
| TEXT | CHARACTER | Set to a character string |
| NOERR | INTEGER | Returns an error number |
This subroutine can be used to write user-defined textual output to the Fortran units associated with 'LIST' and 'ERROR'.
The output of subroutines such as TQCEL, TQMAPL, and TQSHOW, as well as ChemApp error messages can be redirected to files, using TQCIO. Unless the application program is written in Fortran and ChemApp is used as a static library, it is difficult or even impossible to write arbitrary user-defined text to the same file, since it needs to be addressed under a specific Fortran unit number.
For this purpose, TQWSTR may be used, which allows a character string to be written to the Fortran units associated with 'LIST' and 'ERROR'.
The maximum length of the string is determined by the character length conventions of the language in which the application program is written. A length of 255 characters should pose no problem for any application language.
! Redirecting ChemApp output and user-defined text to the same file PROGRAM CAF32 IMPLICIT NONE INTEGER NOERR, FUNIT, LUNIT, IPC, NUMCON CHARACTER LOGTXT*80 DOUBLE PRECISION CO2AMT, DARR2(2) ! Initialise ChemApp CALL TQINI(NOERR) ! Get the unit number from which the thermochemical data-file ! will be read CALL TQGIO('FILE ', FUNIT, NOERR) ! Open the thermochemical data-file cosi.dat (system C-O-Si) ! for reading CALL TQOPNA('cosi.dat', FUNIT, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(FUNIT, NOERR) ! Input 3 mol of CO2/GAS/ CO2AMT = 3.D0 CALL TQINPC('CO2', 1, IPC, NOERR) CALL TQSETC('IA ', 1, IPC, CO2AMT, NUMCON, NOERR) ! Redirect 'LIST' output (e.g. output from TQCEL) to a different ! unit number LUNIT = 21 CALL TQCIO('LIST ', LUNIT, NOERR) ! Open a file under this unit number CALL TQOPEN('caf32.rst', LUNIT, NOERR) ! Write a descriptive text to file 'result' CALL TQWSTR('LIST ', 'Output from TQCEL (ChemSage result table)', * NOERR) 100 FORMAT(A,G8.3) WRITE(LOGTXT, FMT=100) 'Incoming amount of CO2 in mol: ', CO2AMT CALL TQWSTR('LIST ', LOGTXT, NOERR) ! Write ChemSage result table to file 'result' CALL TQCEL(' ',0,0,DARR2,NOERR) ! Direct 'LIST' output back to the standard output unit CALL TQCIO('LIST ', 6, NOERR) ! Close the file 'result' CALL TQCLOS(LUNIT, NOERR) END
/* Program cac32 */ /* Redirecting ChemApp output and user-defined text to the same file */ #include "cacint.h" int main() { LI noerr, funit, lunit, ipc, numcon; char logtxt[80]; DB co2amt, darray2[2]; /* Initialise ChemApp */ tqini(&noerr); /* Get the unit number from which the thermochemical data-file will be read */ tqgio("file", &funit, &noerr); /* Open the thermochemical data-file cosi.dat (system C-O-Si) for reading */ tqopna("cosi.dat", funit, &noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(funit,&noerr); /* Input 3 mol of CO2/GAS/ */ co2amt = 3.0; tqinpc("CO2", 1, &ipc, &noerr); tqsetc("ia ", 1, ipc, co2amt, &numcon, &noerr); /* Redirect "list" output (e.g. output from tqcel) to a different unit number */ lunit = 21; tqcio("list", lunit, &noerr); /* Open a file under this unit number */ tqopen("cac32.rst", lunit, &noerr); /* Write a descriptive text to file "result" */ tqwstr("list", "Output from tqcel (ChemSage result table)", &noerr); sprintf(logtxt, "Incoming amount of CO2 in mol: %li", co2amt); tqwstr("list", logtxt, &noerr); /* Write ChemSage result table to file "result" */ tqcel(" ", 0, 0, darray2, &noerr); /* Direct "list" output back to the standard output unit */ tqcio("list", 6, &noerr); /* Close the file "result" */ tqclos(lunit, &noerr); return 0; }
2.20 TQOPNA
OPEN-ASCII-DATA-FILE
Use TQOPNA to open a thermochemical data-file in ASCII format for reading by ChemApp.
Added for ChemApp version 3.3.0
Synopsis
- FORTRAN:
- CALL TQOPNA(NAME,UNIT,NOERR)
- C:
- tqopna(name,unit,&noerr);
- Pascal:
- tqopna(name,unit,noerr);
- Basic:
- `` Call tqopna(name,unit,noerr)``
| Name | Type | Value set on call or returned |
|---|---|---|
| NAME | CHARACTER | Set to the filename of the ASCII data-file to open |
| UNIT | CHARACTER | Set to the unit number under which it should be opened |
| NOERR | INTEGER | Returns an error number |
Use TQOPNA to open a thermochemical data-file in ASCII format for reading. If you are using ChemApp V3.3.0 or above, it is recommended that you use TQOPNA instead of TQOPEN. The advantage of TQOPNA over TQOPEN is that TQOPNA only opens the data-file if it already exists, otherwise it reports Error Code 107.
The unit number under which the data-file needs to be opened should normally be determined in advance using a call to TQGIO with the option 'FILE'.
A file that has been opened using TQOPNA should subsequently be closed only with TQCLOS.
FORTRAN programmers may use the standard OPEN statement instead of TQOPNA, if ChemApp is used as a static library.
TQOPNA cannot be used to open binary thermochemical data-files. For this purpose, use TQOPNB instead. To open ASCII files for writing, for instance to store the output of TQCEL, TQMAPL, and TQSHOW, use TQOPEN instead.
Please also see Thermodynamic data for details and differences of thermochemical data-files formats.
- See
- TQRFIL
2.21 TQOPNB
OPEN-BINARY-DATA-FILE
Use TQOPNB to open a binary thermochemical data-file for reading by ChemApp.
Added for ChemApp version 3.3.0
Synopsis
- FORTRAN:
- CALL TQOPNB(NAME,UNIT,NOERR)
- C:
- tqopnb(name,unit,&noerr);
- Pascal:
- tqopnb(name,unit,noerr);
- Basic:
- Call tqopnb(name,unit,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NAME | CHARACTER | Set to the filename of the binary data-file to open |
| UNIT | CHARACTER | Set to the unit number under which it should be opened |
| NOERR | INTEGER | Returns an error number |
Use TQOPNB to open a thermochemical data-file in binary format for reading. If you are using ChemApp V3.3.0 or above, it is recommended that you use TQOPNB instead of TQOPEN. The advantage of TQOPNB over TQOPEN is that TQOPNB only opens the data-file if it already exists, otherwise it reports Error Code 107.
The unit number under which the data-file needs to be opened should normally be determined in advance using a call to TQGIO with the option 'FILE'.
A file that has been opened using TQOPNB should subsequently be closed only with TQCLOS.
FORTRAN programmers may use the standard OPEN statement instead of TQOPNB, if ChemApp is used as a static library.
TQOPNB cannot be used to open ASCII thermochemical data-files. For this purpose, use TQOPNA instead.
Please see also Thermodynamic data for details and differences of thermochemical data-files formats.
- See
- TQRBIN
2.22 TQOPNT
OPEN-TRANSPARENT-DATA-FILE
Use TQOPNT to open a transparent thermochemical data-file for reading by ChemApp.
Added for ChemApp version 4.0.0
Synopsis
- FORTRAN:
- CALL TQOPNT(NAME,UNIT,NOERR)
- C:
- tqopnt(name,unit,&noerr);
- Pascal:
- tqopnt(name,unit,noerr);
- Basic:
- Call tqopnt(name,unit,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NAME | CHARACTER | Set to the filename of the transparent data-file to open |
| UNIT | CHARACTER | Set to the unit number under which it should be opened |
| NOERR | INTEGER | Returns an error number |
Use TQOPNT to open a thermochemical data-file in transparent format for reading.
The unit number under which the data-file needs to be opened should normally be determined in advance using a call to TQGIO with the option 'FILE'.
A file that has been opened using TQOPNT should subsequently be closed only with TQCLOS.
FORTRAN programmers may use the standard OPEN statement instead of TQOPNT, if ChemApp is used as a static library. This is not recommended though, because the specifiers needed in calls to OPEN when opening transparent data-files are compiler-dependent.
TQOPNT cannot be used to open ASCII or binary thermochemical data-files. For this purpose, use TQOPNA/TQOPNB instead.
Please see also Thermodynamic data for details and differences of thermochemical data-files formats.
- See
- TQRCST
2.23 TQCLOS
CLOSE-FILE
Use TQCLOS to close a file that has been previously opened using TQOPNA, TQOPNB, TQOPNT, or TQOPEN.
Added for ChemApp version 4.0.0
Synopsis
- FORTRAN:
- CALL TQCLOS(UNIT,NOERR)
- C:
- tqclos(unit,&noerr);
- Pascal:
- tqclos(unit,noerr);
- Basic:
- Call tqclos(unit,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| UNIT | CHARACTER | Set to the unit number under which the data-file was previously opened using TQOPEN |
| NOERR | INTEGER | Returns an error number |
TQCLOS is used to make sure that a file previously opened with TQOPNA, TQOPNB, TQOPNT, or TQOPEN is closed correctly through its associated FORTRAN unit number.
FORTRAN programmers may use the standard CLOSE statement instead of TQCLOS, if ChemApp is used as a static library.
- See
- TQRFIL
2.24 TQGTRH
GET-TRANSPARENT-FILE-HEADER-INFO
Use TQGTRH to retrieve information stored in the header of a transparent file.
Added for ChemApp version 4.0.0
Synopsis
- FORTRAN:
- CALL TQGTRH(VER,NWP,VNW,NRP,VNR,DTC,DTE,ID,USR,REM,NOERR)
- C:
- tqgtrh(&ver,nwp,&vnw,nrp,&vnr,&dtc,&dte,id,usr,rem,&noerr);
- Pascal:
- tqgtrh(ver,nwp,vnw,nrp,vnr,dtc,dte,id,usr,rem,noerr);
- Basic:
- Call tqgtrh(ver,nwp,vnw,nrp,vnr,dtc,dte,id,usr,rem,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| VER | INTEGER | Returns the version number of the transparent file header format |
| NWP | CHARACTER | Returns the name of the program which wrote the data-file (40 chars) |
| VNW | Array of 3 of INTEGER | Returns the version number of the program which wrote the data-file |
| NRP | CHARACTER | Returns the ID(s) of the programs which are permitted to read the data-file (40 chars) |
| VNR | Array of 3 of INTEGER | Returns the minimum version number of the reading program necessary to load the data-file |
| DTC | Array of 6 of INTEGER | Returns the creation date of the data-file |
| DTE | Array of 6 of INTEGER | Retuns the expiry date of the data-file |
| ID | CHARACTER | Returns the user ID(s) of the license holder(s) who are permitted to read this data-file (255 chars) |
| USR | CHARACTER | Returns the name(s) of the license holder(s) who are permitted to read this data-file (80 chars) |
| REM | CHARACTER | Returns the remark (80 chars) |
| NOERR | INTEGER | Returns an error number |
Note that the lengths of the various character strings returned by TQGTRH are greater than regular ChemApp character strings, which is 24. It needs to be made sure that the character variables in the application program which are used with TQGTRH are of sufficient length. Their required lengths are indicated in the table above.
As mentioned in Thermodynamic data, transparent data-files all have a header which precedes the actual thermochemical data. This header contains a number of fields which can be retrieved using TQGTRH:
| VER: | This single integer indicates the version number of the transparent file header format. It can usually be ignored by the programmer. |
|---|---|
| NWP: | This string (40 characters long) contains the name of the program that wrote the data-file. It can consist of one of the 4-letter identifiers used in the NRP field below, or a real name like "FactSage". |
| VNW: | This array of 3 integers is used to pass the version number of the program that wrote the data-file. In the case of FactSage 5.0, the three integers in this array would be 5, 0, and 0. |
| NRP: | This string (40 characters long) contains one or more program IDs and is used to determine which programs are capable and authorized to read the data-file. The program IDs used are 4-letter mnemonic codes that identify a particular program or library (i.e. ChemApp). For instance, if a data-file is meant to be used with the full version of ChemApp, NRP would be set to CAFU, if it is intended for ChemApp light instead, it would be set to CALI. NRP can also contain more than one program IDs, and also program IDs which contain wildcards, in order to make the data-file available to more than one kind of program or library. This field can usually be ignored by the programmer, as ChemApp itself determines whether it is able and authorized to read a particular data-file. |
| VNR: | If applicable, this array of 3 integers contains the required minimum version number of the program that the data-file is intended for. If, for instance, a data-file contains thermochemical data for a mixture phase that requires a solution model only present in ChemApp version V3.5.0 and later, the three integers would be 3, 5, and 0. This array can usually be ignored by the programmer, as ChemApp itself determines whether it is able to read the data-file based on this version number. If the first integer has a value of -1, NRP can be ignored altogether. |
| DTC: | This array of 6 integers contains the creation date and time of the data-file. The 6 integers indicate the year, month, day, hour, minute, and second of the creation of the data-file. Note that this information is independent of the particular time stamp of the data-file as reported by the operating system. |
| DTE: | The 6 integers in this array (same format as DTC) indicate the expiry date of the data-file. |
| ID: | This string (255 characters long) contains one or more user IDs and is used to determine whether ChemApp is authorized to load the data-file. There are three typical scenarios as to what ID contains and how it is interpreted:
|
| USR: | This string (80 characters long) contains the user name associated with the user ID stored in the previous field (ID). Other than THFID, it is not used to for authorisation checks, but only for information purposes. |
| REM: | This string (80 characters long) contains text that the program writing the transparent data-file may use to store any kind of remark. |
Note that in order for TQGTRH to be able to access and retrieve the various transparent file header fields, a transparent data-file needs to be read successfully first.
- See also
- TQOPNT, TQRCST, Thermodynamic data
- See
- TQRCST
2.25 TQGSU
GET-SYSTEM-UNIT
Use TQGSU to find out what unit is currently in use for a specified quantity.
Synopsis
- FORTRAN:
- CALL TQGSU(OPTION,UNIT,NOERR)
- C:
- tqgsu(option,unit,&noerr);
- Pascal:
- tqgsu(option,unit,noerr);
- Basic:
- Call tqgsu(option,unit,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table Units permitted in ChemApp |
| UNIT | CHARACTER | Returns a string as shown in Table Units permitted in ChemApp |
| NOERR | INTEGER | Returns an error number |
OPTION is character input identifying the quantity. UNIT is character output with values as shown in Table Units permitted in ChemApp . The quantities and units available are those also available in FactSage.
- See also
- TQCSU
! Get and display the system units PROGRAM CAF8 IMPLICIT NONE INTEGER NOERR CHARACTER UNIT*24 ! Initialise ChemApp CALL TQINI(NOERR) ! Get default system units WRITE(UNIT=*,FMT='(A)') 'Default system units:' CALL TQGSU('Pressure ',UNIT,NOERR) WRITE(UNIT=*,FMT='(A)') 'Pressure unit: ' // UNIT CALL TQGSU('Volume ',UNIT,NOERR) WRITE(UNIT=*,FMT='(A)') 'Volume unit: ' // UNIT CALL TQGSU('Temperature ',UNIT,NOERR) WRITE(UNIT=*,FMT='(A)') 'Temperature unit: ' // UNIT CALL TQGSU('Energy ',UNIT,NOERR) WRITE(UNIT=*,FMT='(A)') 'Energy unit: ' // UNIT CALL TQGSU('Amount ',UNIT, NOERR) WRITE(UNIT=*,FMT='(A)') 'Amount unit: ' // UNIT END
Default system units: Pressure unit: bar Volume unit: dm3 Temperature unit: K Energy unit: J Amount unit: mol
/* Program cac8 */ /* Get and display the system units */ #include "cacint.h" int main() { LI noerr; char unit[TQSTRLEN]; /* Initialise ChemApp */ tqini(&noerr); /* Get default system units */ printf("Default system units:\n"); tqgsu("Pressure",unit,&noerr); printf("Pressure unit: %s\n", unit); tqgsu("Volume",unit,&noerr); printf("Volume unit: %s\n", unit); tqgsu("Temperature",unit,&noerr); printf("Temperature unit: %s\n", unit); tqgsu("Energy",unit,&noerr); printf("Energy unit: %s\n", unit); tqgsu("Amount",unit, &noerr); printf("Amount unit: %s\n", unit); return 0; }
Default system units: Pressure unit: bar Volume unit: dm3 Temperature unit: K Energy unit: J Amount unit: mol
2.26 TQCSU
CHANGE-SYSTEM-UNIT
Use TQCSU to change the default or selected system unit for a specified quantity.
Synopsis
- FORTRAN:
- CALL TQCSU(OPTION,UNIT,NOERR)
- C:
- tqcsu(option,unit,&noerr);
- Pascal:
- tqcsu(option,unit,noerr);
- Basic:
- Call tqcsu(option,unit,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table Units permitted in ChemApp |
| UNIT | CHARACTER | Returns a string as shown in Table Units permitted in ChemApp |
| NOERR | INTEGER | Returns an error number |
OPTION and UNIT are both character inputs identifying a quantity and a unit, respectively. The selected units remain as they are until changed again using TQCSU, or reset to their defaults using TQINI or TQREMC. The first unit mentioned within each group in Table Units permitted in ChemApp is the default unit for a quantity.
- See also
- TQGSU
! Change system units PROGRAM CAF9 IMPLICIT NONE INTEGER NOERR ! Initialise ChemApp CALL TQINI(NOERR) ! Change system units ! Changing pressure unit to torr CALL TQCSU('Pressure ','torr ', NOERR) ! Changing volume unit to cubic feet CALL TQCSU('Volume ','ft3 ', NOERR) ! Changing temperature unit to Fahrenheit CALL TQCSU('Temperature ','F ', NOERR) ! Changing energy unit to Btu CALL TQCSU('Energy ','Btu ', NOERR) ! Changing amount unit to pound CALL TQCSU('Amount ','pound ', NOERR) END
/* Program cac9 */ /* Change system units */ #include "cacint.h" int main() { LI noerr; /* Initialise ChemApp */ tqini(&noerr); /* Change units */ /* Changing pressure unit to torr */ tqcsu("Pressure","torr", &noerr); /* Changing volume unit to cubic feet */ tqcsu("Volume","ft3", &noerr); /* Changing temperature unit to Fahrenheit */ tqcsu("Temperature","F", &noerr); /* Changing energy unit to Btu */ tqcsu("Energy","Btu", &noerr); /* Changing amount unit to pound */ tqcsu("Amount","pound", &noerr); return 0; }
| Quantity | Unit | Comment |
|---|---|---|
| Pressure | bar | P/bar |
| atm | P/atm = P/bar/1.01325 | |
| Pa | P/Pa = P/bar/0.00001 | |
| kPa | P/kPa = P/bar/0.01 | |
| psi | P/psi = P/bar/0.06894757 | |
| torr | P/torr = P/bar x 1.01325/760 | |
| Volume | dm3 | V/dm3 |
| cm3 | V/cm3 = V/dm3/0.001 | |
| m3 | V/m3 = V/dm3/1000 | |
| ft3 | V/ft3 = V/dm3/28.316846592 | |
| in3 | V/in3 = V/dm3/0.016387064 | |
| Temperature | K | T/K |
| C | T/C = (T/K - 273.15) | |
| F | T/F = 1.8 x (T/K - 273.15)+32 | |
| Energy | J | E/J |
| cal | E/cal = E/J/4.184 | |
| Btu | E/Btu = E/J/1055.06 | |
| kWh | E/kWh = E/J/3,600,000 | |
| Amount | mol | M/mol |
| gram | M/gram | |
| kg | M/gram/1,000 | |
| tonne | M/gram/1,000,000 | |
| pound | M/gram/453.59237 |
3 Handling System Data in ChemApp
Two aspects related to system data have to be considered when programming ChemApp:
- Identifying phases, phase constituents, and system components.
- Optionally setting the status of phases and/or constituents from entered to eliminated or deleted in a calculation.
3.1 Identifying phases, constituents, and components
In ChemApp, phases, phase constituents, and system components in a thermodynamic data-file are automatically numbered sequentially (see also Definition of terms). Phases are numbered in the order gas, condensed mixtures, and finally stoichiometric condensed phases. Phase constituents are numbered starting from one for each phase.
Internally, ChemApp handles phases, phase constituents, and system components using these index numbers, not their names. For this purpose, ChemApp provides a number of subroutines which let you determine the index number associated with a name, and vice versa. Most routines in this group fall into this category.
The total number of components and phases in the system, or constituents in a phase can be obtained by calling TQNOSC, TQNOP, and TQNOPC, respectively. The index number of each system component, phase, and phase constituent can be found by calling TQINSC, TQINP, and TQINPC, and these numbers can be converted back to corresponding names using TQGNSC, TQGNP, and TQGNPC, respectively.
TQSTSC retrieves the stoichiometry of a system component, while TQSTPC does the same for a phase constituent. TQMODL gets the model name of a phase, and TQPCIS checks whether a phase constituent is permitted to be used as an incoming species. TQCSC is used to define a new set of system components.
ChemApp provides four subroutines that retrieve information about sublattices and sublattice constiuents of phases that are modelled according to the sublattice formalism. TQINLC gets the index number of a specified sublattice constituent name, whereas TQGNLC does the opposite, it gets the name for a specified sublattice constituent from its index number. TQNOSL retrieves the number of sublattices for a specified phase, and TQNOLC the number of sublattice constituents for a specified sublattice.
The ChemApp subroutines for obtaining index numbers and other system data are presented in Table ChemApp subroutines for identifying system data.
| Subroutine | Function |
|---|---|
| TQINSC | Gets the index number for a system component |
| TQGNSC | Gets the name for a system component |
| TQCNSC | Changes the name of a system component |
| TQNOSC | Gets the number of system components |
| TQSTSC | Gets the stoichiometry of a system component |
| TQCSC | Changes the system components |
| TQINP | Gets the index number for a phase |
| TQGNP | Gets the name for a phase |
| TQMODL | Gets the model name for a phase |
| TQNOP | Gets the number of phases |
| TQINPC | Gets the index number for a phase constituent |
| TQGNPC | Gets the name for a phase constituent |
| TQPCIS | Checks if a phase constituent is permitted as incoming species |
| TQNOPC | Gets the number of phase constituents |
| TQSTPC | Gets the stoichiometry of a phase constituent |
| TQCHAR | Gets the charge of a phase constituent |
| TQINLC | Gets the index number for a sublattice constituent |
| TQGNLC | Gets the name for a sublattice constituent |
| TQNOSL | Gets the number of sublattices |
| TQNOLC | Gets the number of sublattice constituents |
3.2 TQINSC
GET-INDEX-NUMBER-OF-SYSTEM-COMPONENT
Use TQINSC to get the index number of a specified system component.
Synopsis
- FORTRAN:
- CALL TQINSC(NAME,INDEXS,NOERR)
- C:
- tqinsc(name,&indexs,&noerr);
- Pascal:
- tqinsc(name,indexs,noerr);
- Basic:
- Call tqinsc(name,indexs,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NAME | CHARACTER | Set to a component name |
| INDEXS | INTEGER | Returns the index number for the component |
| NOERR | INTEGER | Returns an error number |
The input variable NAME is permitted to have a maximum length of 24 characters. The returned value for INDEXS is normally used as input to other ChemApp subroutines. The index number stays the same for a specific component as long as the system components remain unchanged (see TQCSC).
! Handling system components PROGRAM CAF10 IMPLICIT NONE INTEGER NOERR, NSCOM, I CHARACTER FNAME*12, SCNAME*24 ! Initialise ChemApp CALL TQINI(NOERR) FNAME = 'cosi.dat' ! Open the thermochemical data-file cosi.dat (system C-O-Si) ! for reading CALL TQOPNA(FNAME, 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Get the number of system components CALL TQNOSC(NSCOM, NOERR) WRITE(*,FMT='(A,I3)') 'Number of system components ' // * 'in file ' // FNAME // ':', NSCOM
Number of system components in file cosi.dat : 3
! Print the names of all system components WRITE(*,FMT='(A)') 'Names of system components:' DO I=1, NSCOM CALL TQGNSC(I, SCNAME, NOERR) WRITE(*,FMT='(I2,A)') I, ': ' // SCNAME ENDDO
Names of system components: 1: C 2: O 3: Si
! Change the names of the system components CALL TQCNSC(1, 'Carbon ', NOERR) CALL TQCNSC(2, 'Oxygen ', NOERR) CALL TQCNSC(3, 'Silicon ', NOERR) ! Print the new names of all system components WRITE(*,FMT='(A)') 'New names of system components:' DO I=1, NSCOM CALL TQGNSC(I, SCNAME, NOERR) WRITE(*,FMT='(I2,A)') I, ': ' // SCNAME ENDDO
New names of system components: 1: Carbon 2: Oxygen 3: Silicon
! Get the index number of a system component CALL TQINSC('Carbon ', I, NOERR) WRITE(*,FMT='(A,I2,A)') 'Carbon is system component no. ', * I, ' in file ' // FNAME END
Carbon is system component no. 1 in file cosi.dat
/* Program cac10 */ /* Handling system components */ #include "cacint.h" int main() { LI noerr, nscom, i; char fname[13], scname[TQSTRLEN]; /* Initialise ChemApp */ tqini(&noerr); strcpy(fname,"cosi.dat"); /* Open the thermochemical data-file cosi.dat (system C-O-Si) for reading */ tqopna(fname,10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Get the number of system components */ tqnosc(&nscom, &noerr); printf("Number of system component in file %s: %li\n", fname, nscom);
Number of system component in file cosi.dat: 3
/* Print the names of all system components */ printf("Names of system components:\n"); for (i = 1; i <= nscom; i++) { tqgnsc(i, scname, &noerr); printf("%li: %s\n", i, scname); }
Names of system components: 1: C 2: O 3: Si
/* Change the names of the system components */ tqcnsc(1, "Carbon", &noerr); tqcnsc(2, "Oxygen", &noerr); tqcnsc(3, "Silicon", &noerr); /* Print the new names of all system components */ printf("New names of system components:\n"); for (i = 1; i <= nscom; i++) { tqgnsc(i, scname, &noerr); printf("%li: %s\n", i, scname); }
New names of system components: 1: Carbon 2: Oxygen 3: Silicon
/* Get the index number of a system component */ tqinsc("Carbon", &i, &noerr); printf("Carbon is system component no. %li in file %s\n", i, fname); return 0; }
Carbon is system component no. 1 in file cosi.dat
3.3 TQGNSC
GET-NAME-OF-SYSTEM-COMPONENT
Use TQGNSC to get the name for a specified system component.
Synopsis
- FORTRAN:
- CALL TQGNSC(INDEXS,NAME,NOERR)
- C:
- tqgnsc(indexs,name,&noerr);
- Pascal:
- tqgnsc(indexs,name,noerr);
- Basic:
- Call tqgnsc(indexs,name,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXS | INTEGER | Set to the index number for a component |
| NAME | CHARACTER | Returns the component name |
| NOERR | INTEGER | Returns an error number |
INDEXS is integer input. The output variable NAME cannot exceed a maximum length of 24 characters.
- See
- TQINSC
3.4 TQCNSC
CHANGE-NAME-OF-SYSTEM-COMPONENT
Use TQCNSC to change the name of a specified system component.
Added for ChemApp version 5.4.4
Synopsis
- FORTRAN:
- CALL TQCNSC(INDEXS,NAME,NOERR)
- C:
- tqcnsc(indexs,name,&noerr);
- Pascal:
- tqcnsc(indexs,name,noerr);
- Basic:
- Call tqcnsc(indexs,name,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXS | INTEGER | Set to the index number for a system component |
| NAME | CHARACTER | Set to the new system component name |
| NOERR | INTEGER | Returns an error number |
INDEXS is integer input. The input character variable NAME cannot exceed a maximum length of 24 characters.
- See
- TQINSC
3.5 TQNOSC
GET-NUMBER-OF-SYSTEM-COMPONENTS
Use TQNOSC to get the total number of system components.
Synopsis
- FORTRAN:
- CALL TQNOSC(NSCOM,NOERR)
- C:
- tqnosc(&nscom,&noerr);
- Pascal:
- tqnosc(nscom,noerr);
- Basic:
- Call tqnosc(nscom,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NSCOM | INTEGER | Returns the total number of components |
| NOERR | INTEGER | Returns an error number |
NSCOM is normally used as an upper limit in loops over all system components. Note that NSCOM may be different from the total number of chemical elements in the system. It can be less when compounds are chosen as components, and greater when the system includes phase-internal electrons.
- See
- TQINSC
3.6 TQSTSC
GET-STOICHIOMETRY-OF-SYSTEM-COMPONENT
Use TQSTSC to get the stoichiometry and molecular mass of a system component.
Synopsis
- FORTRAN:
- CALL TQSTSC(INDEXS,STOI,WMASS,NOERR)
- C:
- tqstsc(indexs,&stoi,&wmass,&noerr);
- Pascal:
- tqstsc(indexs,stoi,wmass,noerr);
- Basic:
- Call tqstsc(indexs,stoi,wmass,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXS | INTEGER | Set to the index number for a component |
| STOI | Array of DOUBLE PRECISION | Returns the stoichiometry of the component |
| WMASS | DOUBLE PRECISION | Returns the molecular mass of the component |
| NOERR | INTEGER | Returns an error number |
INDEXS is integer input. The array STOI needs to be declared large enough to hold at least as much DOUBLE PRECISION values as there are system components, see parameter NSCOM in TQNOSC. The component's full stoichiometry matrix is always returned through STOI, even if known from the component's index number. WMASS is the molecular mass of a component with index number INDEXS.
The contents of the array STOI are fairly trivial: The array is filled with zeros except for the element no. INDEXS, which has a value of 1.0. The reason for TQSTSC to return this array despite its redundant contents is to keep its calling syntax similar to that of TQSTPC. For more information on the stoichiometry matrix, see Definition of a stoichiometry matrix.
Note that the value for the molecular mass is returned in the current amount unit per mol. Since the default amount unit is also mol (see Table Units permitted in ChemApp), the result will always be 1.0, unless the amount unit is changed, for instance to gram.
- See
- TQCSC
3.7 TQCSC
CHANGE-SYSTEM-COMPONENTS
Use TQCSC to define a new set of system components.
Synopsis
- FORTRAN:
- CALL TQCSC(NAME,NOERR)
- C:
- tqcsc(name,&noerr);
- Pascal:
- tqcsc(name,noerr);
- Basic:
- Call tqcsc(name,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NAME | Vector (1-dim. array) of CHARACTER | Set to component names |
| NOERR | INTEGER | Returns an error number |
NAME is a character array input including the NSCOM names (see TQNOSC) of the new set of linearly independent system components. The array may contain names of previous components and of phase constituents, each with a maximum length of 24 characters. The new components are numbered in the order in which they are supplied in this call. Note that NSCOM can be different from the total number of chemical elements in the system. Components may include, for example, oxides only, or phase-internal electrons.
Note that defining a new set of system components is not possible for a system that contains one or more phases modeled using the SUBO (Two-sublattice order/disorder) formalism.
! Changing system components PROGRAM CAF11 IMPLICIT NONE INTEGER NOERR, ISIO2 CHARACTER SCNAME*24, NEWSC(3)*24 DOUBLE PRECISION WMASS, STOI(3) ! Initialise ChemApp CALL TQINI(NOERR) ! Open the thermochemical data-file cosi.dat (system C-O-Si) ! for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Set amount unit to gram, so we get the molecular mass in g/mol CALL TQCSU('Amount ', 'gram ', NOERR) ! Get the name of the first system component CALL TQGNSC(1, SCNAME, NOERR) ! Get its molecular mass CALL TQSTSC(1,STOI,WMASS,NOERR) WRITE(*,FMT='(A)') 'Name of first system component: ' // SCNAME WRITE(*,FMT='(A,G12.5)') 'Molecular mass in g/mol: ', WMASS
Name of first system component: C Molecular mass in g/mol: 12.011
! Get the stoichiometry matrix of SiO2(quartz) CALL TQINP('SiO2(quartz) ',ISIO2,NOERR) CALL TQSTPC(ISIO2,1,STOI,WMASS,NOERR) ! Show the stoichiometry matrix of SiO2(quartz) WRITE(*,'(A)') 'Stoichiometry matrix of SiO2 ' // * 'in terms of system components C-O-Si:' WRITE(*,'(3G12.5)') STOI
Stoichiometry matrix of SiO2 in terms of system components C-O-Si: 0.0000 2.0000 1.0000
! Change system components from C-O-Si to SiO-Si2C-CO2. Note that ! SiO-Si2C-CO2 form a system of three linearly independent components, ! just as the three elements C-O-Si do. NEWSC(1) = 'SiO' NEWSC(2) = 'Si2C' NEWSC(3) = 'CO2' CALL TQCSC(NEWSC,NOERR) ! Now get the name and molecular mass of the first system ! component again WRITE(*,FMT='(A)') 'After changing the system components:' ! Get the name of the first system component CALL TQGNSC(1, SCNAME, NOERR) ! Get its molecular mass CALL TQSTSC(1,STOI,WMASS,NOERR) WRITE(*,FMT='(A)') 'Name of first system component: ' // SCNAME WRITE(*,FMT='(A,G12.5)') 'Molecular mass in g/mol: ', WMASS
After changing the system components: Name of first system component: SiO Molecular mass in g/mol: 44.085
! Get and display the new stoichiometry matrix for SiO2(quartz): CALL TQSTPC(ISIO2,1,STOI,WMASS,NOERR) WRITE(*,'(A)') 'Stoichiometry matrix of SiO2 ' // * 'in terms of system components SiO-Si2C-CO2:' WRITE(*,'(3G12.5)') STOI END
Stoichiometry matrix of SiO2 in terms of system components SiO-Si2C-CO2: 1.5000 -0.25000 0.25000
/* Program cac11 */ /* Changing system components */ #include "cacint.h" int main() { LI noerr, iSiO2; char scname[TQSTRLEN], newsc[4][TQSTRLEN]; DB wmass, stoi[3]; /* Initialise ChemApp */ tqini(&noerr); /* Open the thermochemical data-file cosi.dat (system C-O-Si) for reading */ tqopna("cosi.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Set amount unit to gram, so we get the molecular mass in g/mol */ tqcsu("Amount", "gram", &noerr); /* Get the name of the first system component */ tqgnsc(1, scname, &noerr); /* Get its molecular mass */ tqstsc(1, stoi, &wmass, &noerr); printf("Name of first system component: %s\n", scname); printf("Molecular mass in g/mol: %g\n", wmass);
Name of first system component: C Molecular mass in g/mol: 12.011
/* Get the stoichiometry matrix of SiO2(quartz) */ tqinp("SiO2(quartz)",&iSiO2,&noerr); tqstpc(iSiO2,1,stoi,&wmass,&noerr); /* Show the stoichiometry matrix of SiO2(quartz) */ printf("Stoichiometry matrix of SiO2 " "in terms of system components C-O-Si:\n"); printf("%6.3f %6.3f %6.3f\n", stoi[0], stoi[1], stoi[2]);
Stoichiometry matrix of SiO2 in terms of system components C-O-Si: 0.000 2.000 1.000
/* Change system components from C-O-Si to SiO-Si2C-CO2. Note that SiO-Si2C-CO2 form a system of three linearly independent components, just as the three elements C-O-Si do. */ /* Note that when using C/C++ in contrast to FORTRAN (see above), the array of chars which is passed to tqcsc ("newsc" below) has to consist of one more string than there are system components, and this one extra string has to be empty. This is needed by ChemApp's C/C++ Interface to properly pass arrays of chars to FORTRAN */ strcpy(newsc[0],"SiO"); strcpy(newsc[1],"Si2C"); strcpy(newsc[2],"CO2"); strcpy(newsc[3],""); tqcsc((CHP)newsc,&noerr); /* Note that an explicit typecast has been used in the above call to tqcsc to avoid potential compiler warnings about newsc being an incompatible pointer type */ /* Now get the name and molecular mass of the first system component again */ printf("After changing the system components:\n"); /* Get the name of the first system component */ tqgnsc(1, scname, &noerr); /* Get its molecular mass */ tqstsc(1, stoi, &wmass, &noerr); printf("Name of first system component: %s\n", scname); printf("Molecular mass in g/mol: %g\n", wmass);
After changing the system components: Name of first system component: SiO Molecular mass in g/mol: 44.0854
/* Get and display the new stoichiometry matrix for SiO2(quartz): */ tqstpc(iSiO2,1,stoi,&wmass,&noerr); printf("Stoichiometry matrix of SiO2 " "in terms of system components SiO-Si2C-CO2:\n"); printf("%6.3f %6.3f %6.3f\n", stoi[0], stoi[1], stoi[2]); return 0; }
Stoichiometry matrix of SiO2 in terms of system components SiO-Si2C-CO2: 1.500 -0.250 0.250
3.8 TQINP
GET-INDEX-NUMBER-OF-PHASE
Use TQINP to get the index number for a specified phase.
Synopsis
- FORTRAN:
- CALL TQINP(NAME,INDEXP,NOERR)
- C:
- tqinp(name,&indexp,&noerr);
- Pascal:
- tqinp(name,indexp,noerr);
- Basic:
- Call tqinp(name,indexp,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NAME | CHARACTER | Set to a phase name |
| INDEXP | INTEGER | Returns the index number for the phase |
| NOERR | INTEGER | Returns an error number |
The input variable NAME cannot exceed 24 characters in length. For a mixture phase that might de-mix and is included twice in the data-file, '#1' and '#2' are automatically added to its name. The value of INDEXP returned is normally used as input to other ChemApp subroutines. The index number of a phase remains the same throughout an application.
! Handling phases PROGRAM CAF12 IMPLICIT NONE INTEGER NOERR, NPHASE, I CHARACTER PNAME*24, MNAME*24 ! Initialise ChemApp CALL TQINI(NOERR) ! Open the thermochemical data-file cosi.dat (system C-O-Si) ! for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Get the number of phases CALL TQNOP(NPHASE, NOERR) WRITE(*,FMT='(A,I3)') 'Number of phases in file cosi.dat: ', * NPHASE
Number of phases in file cosi.dat: 8
! Print the names and models of all phases WRITE(*,FMT='(A)') * 'No. Name of phase Model' DO I=1, NPHASE CALL TQGNP(I, PNAME, NOERR) CALL TQMODL(I, MNAME, NOERR) WRITE(*,FMT='(I3,A30,A6)') I, PNAME, MNAME ENDDO
No. Name of phase Model 1 GAS IDMX 2 C PURE 3 Si PURE 4 SiC PURE 5 SiO2(quartz) PURE 6 SiO2(tridymite) PURE 7 SiO2(cristobali) PURE 8 SiO2(liquid) PURE
! Get the index number of a phase CALL TQINP('SiC ', I, NOERR) WRITE(*,FMT='(A,I2,A)') 'SiC is phase no. ', I, * ' in file cosi.dat' END
SiC is phase no. 4 in file cosi.dat
/* Program cac12 */ /* Handling phases */ #include "cacint.h" int main() { LI noerr, nphase, i; char pname[TQSTRLEN], mname[TQSTRLEN]; /* Initialise ChemApp */ tqini(&noerr); /* Open the thermochemical data-file cosi.dat (system C-O-Si) for reading */ tqopna("cosi.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Get the number of phases */ tqnop(&nphase, &noerr); printf("Number of phases in file cosi.dat: %li\n", nphase);
Number of phases in file cosi.dat: 8
/* Print the names and models of all phases */ printf("No. Name of phase Model\n"); for (i = 1; i <= nphase; i++) { tqgnp(i, pname, &noerr); tqmodl(i, mname, &noerr); printf("%3li %-24s %-6s\n", i, pname, mname); }
No. Name of phase Model 1 GAS IDMX 2 C PURE 3 Si PURE 4 SiC PURE 5 SiO2(quartz) PURE 6 SiO2(tridymite) PURE 7 SiO2(cristobali) PURE 8 SiO2(liquid) PURE
/* Get the index number of a phase */ tqinp("SiC", &i, &noerr); printf("SiC is phase no. %li in file cosi.dat\n", i); return 0; }
SiC is phase no. 4 in file cosi.dat
3.9 TQGNP
GET-NAME-PHASE
Use TQGNP to get the name for a specified phase.
Synopsis
- FORTRAN:
- CALL TQGNP(INDEXP,NAME,NOERR)
- C:
- tqgnp(indexp,name,&noerr);
- Pascal:
- tqgnp(indexp,name,noerr);
- Basic:
- Call tqgnp(indexp,name,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| NAME | CHARACTER | Returns the phase name |
| NOERR | INTEGER | Returns an error number |
INDEXP is integer input. The output variable NAME cannot exceed 24 characters in length.
- See
- TQINP
3.10 TQMODL
GET-MODEL-NAME-OF-PHASE
Use TQMODL to get the model name for a phase
Added for ChemApp version 1.1.3
Synopsis
- FORTRAN:
- CALL TQMODL(INDEXP,NAME,NOERR)
- C:
- tqmodl(indexp,name,&noerr);
- Pascal:
- tqmodl(indexp,name,noerr);
- Basic:
- Call tqmodl(indexp,name,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| NAME | CHARACTER | Returns the model name (see Table Identifiers of phase model names as returned by TQMODL) |
| NOERR | INTEGER | Returns an error number |
TQMODL returns the model name of the phase INDEXP. If the phase INDEXP is not a mixture phase, the string 'PURE' is returned in variable NAME. Table Identifiers of phase model names as returned by TQMODL contains a list of all possible model names.
- See
- TQINP
| Condensed phase models | |
| PURE* | Stoichiometric condensed phase |
| IDMX* | Ideal mixing |
| RKMP | Redlich-Kister-Muggianu polynomial [Red48] |
| QKTO | General polynomial Kohler/Toop formalism [Pel88] |
| SUBL | Compound energy formalism [Sun81] [Zha81] [Hil85] |
| SUBO* | Two-sublattice order/disorder formalism [Lac91] |
| SUBS* | Species chemical potential/bond energy formalism [Oat95] |
| SUBI* | Two-sublattice ionic formalism [Sun91] |
| SUBM | Two-sublattice equivalent fraction formalism [Pel88] |
| SUBE | Extended compound energy formalism [Oat] |
| BDEF* | Binary defect formalism [Pel] |
| QUSL | Two-sublattice equivalent fraction formalism as a polynomial [Des91] |
| GAYE* | Gaye-Kapoor-Frohberg cell formalism with or without non-oxidic solutes [Gay92] |
| QUAS* | Modified quasichemical formalism [Pel86] [Bla87] [Pel93] |
| QSOL* | Modified quasichemical formalism with nonoxidic solutes [Pel86] [Bla87] [Pel93] |
| SUBG | Quadruplet quasichemical model [Cha01] [Pel01] |
| HOCH | Hoch-Arpshofen formalism [Hoc84] [Hoc84b] [Hoc87] |
| WAGN | Unified interaction parameter formalism [Pel86b] |
| Aqueous models | |
| IDWZ* | Ideal aqueous mixing |
| IDDZ* | Davies formalism [Dav62] |
| PIMZ* | Pitzer formalism [Har84] |
| PITZ* | Pitzer formalism with fixed alpha-1 and alpha-2 [Har84] |
| PIWZ* | Pitzer formalism without E-theta and E-theta' [Har84] |
| HELZ* | Revised Helgeson-Kirkham-Flowers formalism [Hel81] |
| HTSZ* | Helgeson-Tanger-Shock formalism (ideal) [Tan88] |
| HTWZ* | Helgeson-Tanger-Shock formalism (Debye-Hückel) [Tan88] |
| HTDZ* | Helgeson-Tanger-Shock formalism (Davies) [Tan88] |
| PIHZ* | Helgeson-Tanger-Shock formalism (Pitzer) [Tan88] |
| SITZ* | Specific ion-interaction formalism [Gre97] |
| Gaseous models | |
| IDVT* | Virial equation with Tsonopoulos second virial coefficient correlation [Tso74] |
| IDBS* | C-H-O-S-N-Ar superfluid formalism [Bel92] |
| User-defined models | |
| USP? | Nonaqueous model with excess partial Gibbs energies supplied by user |
| US?Z* | Aqueous model with excess partial Gibbs energies supplied by user |
| USX? | Model with partial derivatives dG/dx supplied by user |
| USG?* | Gaseous model with second virial coefficients supplied by user |
3.11 TQNOP
GET-NUMBER-OF-PHASES
Use TQNOP to get the total number of phases.
Synopsis
- FORTRAN:
- CALL TQNOP(NPHASE,NOERR)
- C:
- tqnop(&nphase,&noerr);
- Pascal:
- tqnop(nphase,noerr);
- Basic:
- Call tqnop(nphase,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NPHASE | INTEGER | Returns the total number of phases |
| NOERR | INTEGER | Returns an error number |
NPHASE is normally used as upper limit in loops over all phases. A loop over all condensed phases would start from index number 2, since the index number 1 is reserved for the gas phase.
- See
- TQINP
3.12 TQINPC
GET-INDEX-NUMBER-OF-PHASE-CONSTITUENT
Use TQINPC to get the index number for a specified phase constituent.
Synopsis
- FORTRAN:
- CALL TQINPC(NAME,INDEXP,INDEXC,NOERR)
- C:
- tqinpc(name,indexp,&indexc,&noerr);
- Pascal:
- tqinpc(name,indexp,indexc,noerr);
- Basic:
- Call tqinpc(name,indexp,indexc,noerr
| Name | Type | Value set on call or returned |
|---|---|---|
| NAME | CHARACTER | Set to a phase constituent name |
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXC | INTEGER | Returns the index number for the constituent |
| NOERR | INTEGER | Returns an error number |
NAME and INDEXP are input. NAME cannot exceed 24 characters in length. The value of INDEXC returned is normally used as input to other subroutines of the interface. The number remains the same throughout the application.
! Handling phase constituents PROGRAM CAF13 IMPLICIT NONE INTEGER NOERR, NPCON, NSCOM, I CHARACTER PCNAME*24, SCNAME(3)*24 DOUBLE PRECISION WMASS, STOI(3) ! Initialise ChemApp CALL TQINI(NOERR) ! Open the thermochemical data-file cosi.dat (system C-O-Si) ! for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Get the number of phase constituents of the first phase in ! cosi.dat, which is the gaseous mixture phase CALL TQNOPC(1, NPCON, NOERR) WRITE(*,FMT='(A,I3)') 'Number of phase constituents of ' // * 'the gas phase in cosi.dat: ', NPCON
Number of phase constituents of the gas phase in cosi.dat: 15
! Print the names of all phase constituents of the gas phase, ! their stoichiometry in terms of the system components, ! and their molecular mass ! Get names of the system components CALL TQNOSC(NSCOM, NOERR) DO I=1, NSCOM CALL TQGNSC(I, SCNAME(I), NOERR) ENDDO ! Print header WRITE(*,FMT='(A)') ' ' // * 'Stoichiometry Mol. mass' WRITE(*,FMT='(A)') 'No. Name of phase const. ' // * SCNAME(1)(1:6) // * SCNAME(2)(1:6) // * SCNAME(3)(1:6) // * ' g/mol' ! Set amount unit to gram, so we get the molecular mass in g/mol CALL TQCSU('Amount ', 'gram ', NOERR) ! Get the information on all phase constituents of ! the gas phase DO I=1, NPCON CALL TQGNPC(1, I, PCNAME, NOERR) CALL TQSTPC(1, I, STOI, WMASS, NOERR) WRITE(*,FMT='(I3,A26,3F6.1,2X,G12.5)') * I, PCNAME, STOI(1), STOI(2), STOI(3), * WMASS ENDDO
Stoichiometry Mol. mass
No. Name of phase const. C O Si g/mol
1 C 1.0 0.0 0.0 12.011
2 C2 2.0 0.0 0.0 24.022
3 C3 3.0 0.0 0.0 36.033
4 CO 1.0 1.0 0.0 28.010
5 CO2 1.0 2.0 0.0 44.010
6 O 0.0 1.0 0.0 15.999
7 O2 0.0 2.0 0.0 31.999
8 O3 0.0 3.0 0.0 47.998
9 Si 0.0 0.0 1.0 28.086
10 Si2 0.0 0.0 2.0 56.172
11 Si2C 1.0 0.0 2.0 68.183
12 Si3 0.0 0.0 3.0 84.258
13 SiC 1.0 0.0 1.0 40.097
14 SiO 0.0 1.0 1.0 44.085
15 SiO2 0.0 2.0 1.0 60.085
! Get the index number of a phase constituent, ! in this case, CO2 in the gas phase CALL TQINPC('CO2 ', 1, I, NOERR) WRITE(*,FMT='(A,I2,A)') 'CO2 is phase constituent no. ',I, * ' in the gas phase in file cosi.dat' END
CO2 is phase constituent no. 5 in the gas phase in file cosi.dat
/* Program cac13 */ /* Handling phase constituents*/ #include "cacint.h" int main() { LI noerr, npcon, nscom, i; char pcname[TQSTRLEN], scname[3][TQSTRLEN]; DB wmass, stoi[3]; /* Initialise ChemApp */ tqini(&noerr); /* Open the thermochemical data-file cosi.dat (system C-O-Si) for reading */ tqopna("cosi.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Get the number of phase constituents of the first phase in cosi.dat, which is the gaseous mixture phase */ tqnopc(1, &npcon, &noerr); printf("Number of phase constituents of " "the gas phase in cosi.dat: %li\n", npcon);
Number of phase constituents of the gas phase in cosi.dat: 15
/* Print the names of all phase constituents of the gas phase, their stoichiometry in terms of the system components, and their molecular masses */ /* Get names of the system components */ tqnosc(&nscom, &noerr); for (i = 1; i <= nscom; i++) { tqgnsc(i, scname[i-1], &noerr); } /* Print header */ printf(" " "Stoichiometry Mol. mass\n"); printf("No. Name of phase const. %6s%6s%6s g/mol\n", scname[0],scname[1],scname[2]); /* Set amount unit to gram, so we get the molecular mass in g/mol */ tqcsu("Amount", "gram", &noerr); /* Get the information on all phase constituents of the gas phase */ for (i = 1; i <= npcon; i++) { tqgnpc(1, i, pcname, &noerr); tqstpc(1, i, stoi, &wmass, &noerr); printf("%3li %-26s%6.1f%6.1f%6.1f %g\n", i, pcname, stoi[0], stoi[1], stoi[2], wmass); }
Stoichiometry Mol. mass
No. Name of phase const. C O Si g/mol
1 C 1.0 0.0 0.0 12.011
2 C2 2.0 0.0 0.0 24.022
3 C3 3.0 0.0 0.0 36.033
4 CO 1.0 1.0 0.0 28.0104
5 CO2 1.0 2.0 0.0 44.0098
6 O 0.0 1.0 0.0 15.9994
7 O2 0.0 2.0 0.0 31.9988
8 O3 0.0 3.0 0.0 47.9982
9 Si 0.0 0.0 1.0 28.086
10 Si2 0.0 0.0 2.0 56.172
11 Si2C 1.0 0.0 2.0 68.183
12 Si3 0.0 0.0 3.0 84.258
13 SiC 1.0 0.0 1.0 40.097
14 SiO 0.0 1.0 1.0 44.0854
15 SiO2 0.0 2.0 1.0 60.0848
/* Get the index number of a phase constituent, in this case, CO2 in the gas phase */ tqinpc("CO2", 1, &i, &noerr); printf("CO2 is phase constituent no. %li " "in the gas phase in file cosi.dat\n", i); return 0; }
CO2 is phase constituent no. 5 in the gas phase in file cosi.dat
3.13 TQGNPC
GET-NAME-OF-PHASE-CONSTITUENT
Use TQGNPC to get the name for a specified phase constituent.
Synopsis
- FORTRAN:
- CALL TQGNPC(INDEXP,INDEXC,NAME,NOERR)
- C:
- tqgnpc(indexp,indexc,name,&noerr);
- Pascal:
- tqgnpc(indexp,indexc,name,noerr);
- Basic:
- Call tqgnpc(indexp,indexc,name,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXC | INTEGER | Set to the index number for a constituent |
| NAME | CHARACTER | Returns the constituent name |
| NOERR | INTEGER | Returns an error number |
INDEXP and INDEXC are integer input. The output variable NAME cannot exceed 24 characters in length.
- See
- TQINPC
3.14 TQPCIS
CHECK-IF-PHASE-CONSTITUENT-IS-PERMITTED-AS-INCOMING-SPECIES
Use TQPCIS to check if a phase constituent is permitted as incoming species.
Added for ChemApp version 3.1.0
Synopsis
- FORTRAN:
- CALL TQPCIS(INDEXP,INDEXC,INPCIS,NOERR)
- C:
- tqpcis(indexp,indexc,&inpcis,&noerr);
- Pascal:
- tqpcis(indexp,indexc,inpcis,noerr);
- Basic:
- Call tqpcis(indexp,indexc,inpcis,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXC | INTEGER | Set to the index number for a constituent |
| INPCIS | INTEGER | Returns 1 if permitted, 0 if not |
| NOERR | INTEGER | Returns an error number |
INDEXP and INDEXC are input. If the specified phase constituent can be used as an incoming species using TQSETC/TQSTCA, INPCIS returns 1, otherwise it returns 0.
Note that only for the sparsely used solution model SUBS (see Tables List of available solution models in ChemApp and Identifiers of phase model names as returned by TQMODL) can INPCIS take values equal to zero.
! Determining whether phase constituents are permitted as incoming ! species PROGRAM CAF25 IMPLICIT NONE INTEGER NOERR, NPHASE, NPCON, I, J, INPCIS, NNPERM CHARACTER PNAME*24, PCNAME*24 ! Initialise the number of phase constituents which cannot be used as ! incoming species to 0 NNPERM = 0 ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Check whether the data-file contains any phase constituents that ! cannot be used as incoming species. ! Get the number of phases CALL TQNOP(NPHASE, NOERR) 100 FORMAT(1X,A,T26,A,T51,A) ! Print a table header WRITE(*,FMT=100) 'PHASE', 'PHASE CONSTITUENT', * 'O.K./NOT PERMITTED' ! Loop over all phases DO I=1, NPHASE CALL TQGNP(I, PNAME, NOERR) CALL TQNOPC(I, NPCON, NOERR) ! We only need to check mixture phases IF (NPCON .GT. 1) THEN ! Loop over all constituents DO J=1, NPCON CALL TQGNPC(I, J, PCNAME, NOERR) ! Check whether the current constituent is permitted to be used as ! incoming species CALL TQPCIS(I, J, INPCIS, NOERR) ! Print a table entry, and if it is not permitted, increase the total ! number of such phase constituents by 1 IF (INPCIS .EQ. 0) THEN NNPERM = NNPERM + 1 WRITE(*,FMT=100) PNAME, PCNAME, 'not permitted' ELSE WRITE(*,FMT=100) PNAME, PCNAME, 'o.k.' ENDIF ENDDO ENDIF ENDDO ! If there were phase constituents which cannot be used as incoming ! species, print a note. IF (NNPERM .GT. 0) THEN WRITE(*,FMT='(1X,A,I3,A)') 'Note: ', NNPERM, ' phase ' // * 'constituent(s) is/are not permitted ' WRITE(*,FMT='(1X,A)') ' as incoming species.' ENDIF END
PHASE PHASE CONSTITUENT O.K./NOT PERMITTED GAS C o.k. GAS C2 o.k. GAS C3 o.k. GAS CO o.k. GAS CO2 o.k. GAS O o.k. GAS O2 o.k. GAS O3 o.k. GAS Si o.k. GAS Si2 o.k. GAS Si2C o.k. GAS Si3 o.k. GAS SiC o.k. GAS SiO o.k. GAS SiO2 o.k.
/* Program cac25 */ /* Determining whether phase constituents are permitted as incoming species */ #include "cacint.h" int main() { LI noerr, nphase, npcon, i, j, inpcis, nnperm; char pname[TQSTRLEN], pcname[TQSTRLEN]; /* Initialise the number of phase constituents which cannot be used as incoming species to 0 */ nnperm = 0; /* Initialise ChemApp */ tqini(&noerr); /* Open data-file for reading */ tqopna("cosi.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Check whether the data-file contains any phase constituents that cannot be used as incoming species. */ /* Get the number of phases */ tqnop(&nphase, &noerr); /* Print a table header */ printf("%-25s%-25s%-19s\n", "PHASE", "PHASE CONSTITUENT", "O.K./NOT PERMITTED"); /* Loop over all phases */ for (i=1; i<=nphase; i++) { tqgnp(i, pname, &noerr); tqnopc(i, &npcon, &noerr); /* We only need to check mixture phases */ if (npcon > 1) { /* Loop over all constituents */ for (j=1; j<=npcon; j++) { tqgnpc(i, j, pcname, &noerr); /* Check whether the current constituent is permitted to be used as incoming species */ tqpcis(i, j, &inpcis, &noerr); /* Print a table entry, and if it is not permitted, increase the total number of such phase constituents by 1 */ if (inpcis == 0) { nnperm++; printf("%-25s%-25s%-19s\n", pname, pcname, "not permitted"); } else { printf("%-25s%-25s%-19s\n", pname, pcname, "o.k."); } } } } /* If there were phase constituents which cannot be used as incoming species, print a note. */ if (nnperm > 0) { printf("Note: %li phase constituent(s) is/are not permitted\n" " as incoming species.\n", nnperm); } return 0; }
PHASE PHASE CONSTITUENT O.K./NOT PERMITTED GAS C o.k. GAS C2 o.k. GAS C3 o.k. GAS CO o.k. GAS CO2 o.k. GAS O o.k. GAS O2 o.k. GAS O3 o.k. GAS Si o.k. GAS Si2 o.k. GAS Si2C o.k. GAS Si3 o.k. GAS SiC o.k. GAS SiO o.k. GAS SiO2 o.k.
3.15 TQNOPC
GET-NUMBER-OF-PHASE-CONSTITUENTS
Use TQNOPC to get the total number of constituents in a specified phase.
Synopsis
- FORTRAN:
- CALL TQNOPC(INDEXP,NPCON,NOERR)
- C:
- tqnopc(indexp,&npcon,&noerr);
- Pascal:
- tqnopc(indexp,npcon,noerr);
- Basic:
- Call tqnopc(indexp,npcon,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| NPCON | INTEGER | Returns the total number of constituents in a phase |
| NOERR | INTEGER | Returns an error number |
NPCON is integer output normally used as upper limit in loops over all constituents of phase number INDEXP.
- See
- TQINPC
3.16 TQSTPC
GET-STOICHIOMETRY-OF-PHASE-CONSTITUENT
Use TQSTPC to get the stoichiometry and molecular mass of a specified phase constituent.
Synopsis
- FORTRAN:
- CALL TQSTPC(INDEXP,INDEXC,STOI,WMASS,NOERR)
- C:
- tqstpc(indexp,indexc,&stoi,&wmass,&noerr);
- Pascal:
- tqstpc(indexp,indexc,stoi,wmass,noerr);
- Basic:
- Call tqstpc(indexp,indexc,stoi,wmass,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXC | INTEGER | Set to the index number for a constituent |
| STOI | Array of DOUBLE PRECISION | Returns the stoichiometry of the constituent |
| WMASS | DOUBLE PRECISION | Returns the molecular mass of the constituent |
| NOERR | INTEGER | Returns an error number |
INDEXP and INDEXC are integer input. The array STOI needs to be declared large enough to hold at least as much DOUBLE PRECISION values as there are system components, see parameter NSCOM in TQNOSC. The stoichiometry of constituent INDEXC in phase INDEXP returned through STOI depends on the current set of system components. WMASS is the molecular mass of the same phase constituent.
For more information on the stoichiometry matrix, see Definition of a stoichiometry matrix.
Note that the value for the molecular mass is returned in the current amount unit per mol. Since the default amount unit is also mol (see Table Units permitted in ChemApp), the result will always be 1.0, unless the amount unit is changed, for instance to gram.
- See
- TQINPC
3.17 TQCHAR
GET-CHARGE-OF-PHASE-CONSTITUENT
Use TQCHAR to get the charge of a phase constituent.
Added for ChemApp version 5.4.1
Synopsis
- FORTRAN:
- CALL TQCHAR(INDEXP,INDEXC,VAL,NOERR)
- C:
- tqchar(indexp,indexc,&val,&noerr);
- Pascal:
- tqchar(indexp,indexc,val,noerr);
- Basic:
- Call tqchar(indexp,indexc,val,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXC | INTEGER | Set to the index number for a constituent |
| VAL | DOUBLE PRECISION | Returns the charge |
| NOERR | INTEGER | Returns an error number |
INDEXP and INDEXC are integer input. The output variable VAL is of type DOUBLE PRECISION and contains the charge. Except for aqueous solutions, the output will be correct only if the system component name for a phase-specific electron is of the type 'e(PHASE_NAME)'. Use TQCNSC to rename the system component for a phase-specific electron, if necessary.
3.18 TQINLC
GET-INDEX-NUMBER-OF-SUBLATTICE-CONSTITUENT
Use TQINLC to get the index number of a specified sublattice constituent.
Added for ChemApp version 3.3.1
Synopsis
- FORTRAN:
- CALL TQINLC(NAME,INDEXP,INDEXL,INDEXC,NOERR)
- C:
- tqinlc(name,indexp,indexl,&indexc,&noerr);
- Pascal:
- tqinlc(name,indexp,indexl,indexc,noerr);
- Basic:
- Call tqinlc(name,indexp,indexl,indexc,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NAME | CHARACTER | Set to a sublattice constituent name |
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXL | INTEGER | Set to the index number for a sublattice |
| INDEXC | INTEGER | Returns the index number of the sublattice constituent |
| NOERR | INTEGER | Returns an error number |
The input variable NAME is permitted to have a maximum length of 24 characters. INDEXL has to be set to the index number of the sublattice in the phase INDEXP of which NAME is a constituent. The returned value for INDEXC is normally used as input to other ChemApp subroutines and stays the same throughout an application, or until a different thermochemical data-file is loaded. All solution phases are considered to be modelled with one or several sublattices. For phases with one sublattice, the subroutines TQINLC and TQINPC are equivalent.
- See
- TQGNLC
3.19 TQGNLC
GET-NAME-OF-SUBLATTICE-CONSTITUENT
Use TQGNLC to get the name for a specified sublattice constituent.
Added for ChemApp version 3.3.1
Synopsis
- FORTRAN:
- CALL TQGNLC(INDEXP,INDEXL,INDEXC,NAME,NOERR)
- C:
- tqgnlc(indexp,indexl,&indexc,name,&noerr);
- Pascal:
- tqgnlc(indexp,indexl,indexc,name,noerr);
- Basic:
- Call tqgnlc(indexp,indexl,indexc,name,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXL | INTEGER | Set to the index number for a sublattice |
| INDEXC | INTEGER | Set to the index number for a sublattice constituent |
| NAME | CHARACTER | Returns the sublattice constituent name |
| NOERR | INTEGER | Returns an error number |
INDEXP, INDEXL, and INDEXC are all integer input and have to be set to the index numbers of a phase, a sublattice in that phase, and a sublattice constituent on that sublattice. The output variable NAME cannot exceed 25 characters in length. All solution phases are considered to be modelled with one or several sublattices. For phases with one sublattice, the subroutines TQGNLC and TQGNPC are equivalent.
If the variable NAME contains a name for the sublattice constituent like Comp 1, 1 this usually means that the thermochemical data-file used has been converted from ChemSage version 3.x to 4.x format using the utility ChemFile. Sublattice constituent names were not part of the ChemSage V3.x data-file format, thus ChemFile can only use generic names for them if a data-file is converted that contains a sublattice phase. If the data-file in use is in ASCII format, these generic names can be manually edited in the file using a text editor. Alternatively, the programm CSFAP offered by GTT-Technologies can be used for this purpose. In case binary or transparent data-files are used, please contact GTT-Technologies.
! Handling sublattice constituents PROGRAM CAF26 IMPLICIT NONE INTEGER NOERR, NPHASE, NSL, NSLCON, INDEXP, INDEXL, INDEXC, I CHARACTER PNAME*24, CNAME*24 ! Initialise ChemApp CALL TQINI(NOERR) ! Open the thermochemical data-file subl-ex.dat for reading. It contains ! an extract of the system Co-Cr-Fe: the phase SIGMA:30, which is ! modelled according to the sublattice formalism, and the BCC phase, ! described by a Redlich-Kister polynomial. Both phases are each ! included twice, to account for miscibility gaps. CALL TQOPNA('subl-ex.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! The first of the two identical copies of the SIGMA:30 phase, which ! ChemApp calls SIGMA:30#1, will be investigated with respect to the ! number of sublattices, the number of sublattice constituents on each ! sublattice, and the names of the sublattice constituents. ! Get the index number for the phase SIGMA:30#1 PNAME = 'SIGMA:30#1' CALL TQINP(PNAME, INDEXP, NOERR) ! Get the number of sublattices CALL TQNOSL(INDEXP, NSL, NOERR) WRITE(*,*) PNAME, ' has ', NSL, ' sublattices'
SIGMA:30#1 has 3 sublattices
! Loop over all sublattices DO INDEXL=1, NSL ! Get the number of sublattice constituents CALL TQNOLC(INDEXP, INDEXL, NSLCON, NOERR) WRITE(*,*) 'Sublattice ', INDEXL, ' has ', NSLCON, * ' constituents with the following names:' ! Get the name of each sublattice constituent DO INDEXC=1, NSLCON CALL TQGNLC(INDEXP, INDEXL ,INDEXC, CNAME, NOERR) ! The reverse (getting the index number for the name of the sublattice ! constituent just retrieved), is rather superfluous here, and only used ! to demonstrate the call to TQINLC: CALL TQINLC(CNAME, INDEXP, INDEXL, I, NOERR) WRITE(*,*) ' ',I,':', CNAME ENDDO ENDDO END
Sublattice 1 has 2 constituents with the following names:
1 :Co
2 :Fe
Sublattice 2 has 1 constituents with the following names:
1 :Cr
Sublattice 3 has 3 constituents with the following names:
1 :Co
2 :Cr
3 :Fe
/* Program cac26 */ /* Handling sublattice constituents */ #include "cacint.h" int main() { LI noerr, nphase, nsl, nslcon, indexp, indexl, indexc, i; char pname[TQSTRLEN], cname[TQSTRLEN]; /* Initialise ChemApp */ tqini(&noerr); /* Open the thermochemical data-file subl-ex.dat for reading. It contains an extract of the system Co-Cr-Fe: the phase SIGMA:30, which is modelled according to the sublattice formalism, and the BCC phase, described by a Redlich-Kister polynomial. Both phases are each included twice, to account for miscibility gaps. */ tqopna("subl-ex.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10, &noerr); /* The first of the two identical copies of the SIGMA:30 phase, which ChemApp calls SIGMA:30#1, will be investigated with respect to the number of sublattices, the number of sublattice constituents on each sublattice, and the names of the sublattice constituents. */ /* Get the index number for the phase SIGMA:30#1 */ strcpy(pname, "SIGMA:30#1"); tqinp(pname, &indexp, &noerr); /* Get the number of sublattices */ tqnosl(indexp, &nsl, &noerr); printf("%s has %li sublattices\n",pname, nsl);
SIGMA:30#1 has 3 sublattices
/* Loop over all sublattices */ for (indexl = 1; indexl<=nsl; indexl++) { /* Get the number of sublattice constituents */ tqnolc(indexp, indexl, &nslcon, &noerr); printf("Sublattice %li has %li constituents with the following names:\n", indexl, nslcon); /* Get the name of each sublattice constituent */ for (indexc = 1; indexc<=nslcon; indexc++) { tqgnlc(indexp, indexl ,indexc, cname, &noerr); /* The reverse (getting the index number for the name of the sublattice constituent just retrieved), is rather superfluous here, and only used to demonstrate the call to tqinlc: */ tqinlc(cname, indexp, indexl, &i, &noerr); printf(" %li: %s\n", i, cname); } } return 0; }
Sublattice 1 has 2 constituents with the following names: 1: Co 2: Fe Sublattice 2 has 1 constituents with the following names: 1: Cr Sublattice 3 has 3 constituents with the following names: 1: Co 2: Cr 3: Fe
3.20 TQNOSL
GET-NUMBER-OF-SUBLATTICES
Use TQNOSL to get the number of sublattices for a specified phase.
Added for ChemApp version 3.3.1
Synopsis
- FORTRAN:
- CALL TQNOSL(INDEXP,NSUBL,NOERR)
- C:
- tqnosl(indexp,&nsubl,&noerr);
- Pascal:
- tqnosl(indexp,nsubl,noerr);
- Basic:
- Call tqnosl(indexp,nsubl,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| NSUBL | INTEGER | Returns the number of sublattices |
| NOERR | INTEGER | Returns an error number |
While all solution models are considered to be modelled with one or more sublattices, INDEXP is normally set to the index number of a phase modelled according to a sublattice formalism (see TQMODL). The number of sublattices in this phase returned in NSUBL is normally used as upper limit in loops over all sublattices of phase INDEXP.
3.21 TQNOLC
GET-NUMBER-OF-SUBLATTICE-CONSTITUENTS
Use TQNOLC to get the number of sublattice constituents for a specified sublattice.
Added for ChemApp version 3.3.1
Synopsis
- FORTRAN:
- CALL TQNOLC(INDEXP,INDEXL,NSLCON,NOERR)
- C:
- tqnolc(indexp,indexl,&nslcon,&noerr);
- Pascal:
- tqnolc(indexp,indexl,nslcon,noerr);
- Basic:
- Call tqnolc(indexp,indexl,nslcon,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXL | INTEGER | Set to the index number for a sublattice |
| NSLCON | INTEGER | Returns the number of sublattice constituents |
| NOERR | INTEGER | Returns an error number |
INDEXP has to be set to the index number of a solution phase, INDEXL to the index number of a sublattice in that phase. The number of sublattice constituents in that sublattice returned in NSLCON is normally used as upper limit in loops over all sublattices constituents of sublattice INDEXL in phase INDEXP. All solution phases are considered to be modelled with one or several sublattices.
3.22 Changing the status of phases and constituents
ChemApp contains subroutines that enable the status of phases or constituents to be changed; that is, to be either excluded from or re-entered into an equilibrium calculation (see Table ChemApp subroutines for changing the status of phases and constituents). The default status is that stored in the thermodynamic data-file. The status for a phase can be changed implicitly, dependent on the explicit changes of the statuses of its constituents. For example, if all constituents of a phase are made dormant, the phase will also be dormant.
Whereas complete phases can always be set ENTERED, DORMANT or ELIMINATED, some restrictions apply for phase constituents (see the notes for TQCSPC).
A common reason for changing the status of a phase is for instance the desire to calculate a metastable equilibrium (see TQGSP) or to increase the computational speed in systems containing miscibility gaps (see Data-files with preset suppression of species).
| Subroutine | Function |
|---|---|
| TQGSP | Gets the status of a phase |
| TQCSP | Changes the status of a phase |
| TQGSPC | Gets the status of a phase constituent |
| TQCSPC | Changes the status of a phase constituent |
| Option | Comment |
|---|---|
| ENTERED | The phase or constituent is included in the equilibrium calculation. An included phase is not necessarily stable at equilibrium. |
| DORMANT | The phase or constituent is excluded from the mass balances (it will appear with zero amount at equilibrium), but its activity is calculated. If the calculated activity for a phase is greater than one, it would be stable if entered. |
| ELIMINATED | The phase or constituent is ignored in the equilibrium calculation |
3.23 TQGSP
GET-STATUS-OF-PHASE
Use TQGSP to get the status of a specified phase.
Synopsis
- FORTRAN:
- CALL TQGSP(INDEXP,OPTION,NOERR)
- C:
- tqgsp(indexp,option,&noerr);
- Pascal:
- tqgsp(indexp,option,noerr);
- Basic:
- Call tqgsp(indexp,option,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| OPTION | CHARACTER | Returns a string as shown in Table Possible statuses for phases and constituents |
| NOERR | INTEGER | Returns an error number |
INDEXP is integer input and OPTION is character output identifying the status (see Table Possible statuses for phases and constituents).
! Changing the status of phases SUBROUTINE TABLE IMPLICIT NONE INTEGER I, NOERR, NPHASE DOUBLE PRECISION AMOUNT, ACT CHARACTER NAME*24 ! Retrieve and display the activities and equilibrium ! amounts of all phases ! Get number of phases CALL TQNOP(NPHASE, NOERR) ! Print table header write(*,FMT='(A10,17X,A14,2X,A14)') 'Phase name', * 'amount/mol', * 'activity' DO I=1, NPHASE ! Get the phase name CALL TQGNP(I, NAME, NOERR) ! Get its equilibrium amount CALL TQGETR('A ', I, 0, AMOUNT, NOERR) ! Get its activity CALL TQGETR('AC ', I, 0, ACT, NOERR) write(*,FMT='(A24,2X,F14.5,2X,F14.5)') NAME,AMOUNT,ACT ENDDO RETURN END PROGRAM CAF14 IMPLICIT NONE INTEGER NOERR, NUMCON INTEGER ISIC, ISIO2, IC DOUBLE PRECISION VALS(2) CHARACTER STATUS*24 ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Get the phase index number of SiC CALL TQINP('SiC ', ISIC, NOERR) ! Get the status of this phase CALL TQGSP(ISIC, STATUS, NOERR) WRITE(*,FMT='(A)') 'The status of SiC is currently ' // * STATUS
The status of SiC is currently ENTERED
! Unless a phase is marked "dormant" or "eliminated" in the data-file ! itself, the status is by default always "entered". This means it is ! considered in the subsequent equilibrium calculations and is included ! in the mass balance. ! Set some conditions and perform a simple calculation to ! demonstrate the effect of the different statuses ! Set temperature to 1900 K CALL TQSETC('T ', 0, 0, 1900.D0, NUMCON, NOERR) ! Set incoming amount to 1 mol SiO2(quartz) and 2 mol C CALL TQINP('SiO2(quartz)', ISIO2, NOERR) CALL TQSETC('IA ', ISIO2, 0, 1.D0, NUMCON, NOERR) CALL TQINP('C ', IC, NOERR) CALL TQSETC('IA ', IC, 0, 2.D0, NUMCON, NOERR) ! Calculate the equilibrium CALL TQCE(' ', 0, 0, VALS, NOERR) ! Print a table containing the phase names, their ! equilibrium amounts, and activities CALL TABLE
Phase name amount/mol activity GAS 1.39778 1.00000 C 0.00000 0.43860 Si 0.00000 0.07912 SiC 0.65059 1.00000 SiO2(quartz) 0.00000 0.90635 SiO2(tridymite) 0.00000 0.99918 SiO2(cristobali) 0.30105 1.00000 SiO2(liquid) 0.00000 0.97125
! From the above table it can be seen that SiC is stable ! under those conditions. Its activity is unity and its ! equilibrium amount is approx. 0.65 mol ! Now change the status of SiC to "dormant" and rerun the ! calculation CALL TQCSP(ISIC, 'DORMANT ', NOERR) CALL TQCE(' ', 0, 0, VALS, NOERR) CALL TABLE
Phase name amount/mol activity GAS 0.00000 0.74210 C 2.00000 1.00000 Si 0.00000 1.00000 SiC 0.00000 28.81864 SiO2(quartz) 0.00000 0.90635 SiO2(tridymite) 0.00000 0.99918 SiO2(cristobali) 1.00000 1.00000 SiO2(liquid) 0.00000 0.97125
! Setting a phase to "dormant" tells ChemApp to exclude it from the mass ! balance, but still calculate its activity. This offers the ! possibility to consider a phase to be metastable, while still ! obtaining information about its activity. It can be seen from the ! above table that ChemApp sets the equilibrium amount of SiC to zero, ! and calculated its activity to be almost 30. Instead of SiC, ChemApp ! now calculates Si to be stable. ! If a phase is to be completely ignored during the equilibrium ! calculations, its status should be set to "eliminated", as in the ! following example. CALL TQCSP(ISIC, 'ELIMINATED ', NOERR) CALL TQCE(' ', 0, 0, VALS, NOERR) CALL TABLE ! Now ChemApp ignored SiC during the equilibrium calculations, as if it ! were not even included in the data-file. The status "eliminated" ! allows one to selectively delete phases from equilibrium calculations, ! for instance to increase computational efficiency. END
Phase name amount/mol activity GAS 0.00000 0.74210 C 2.00000 1.00000 Si 0.00000 1.00000 SiC 0.00000 0.00000 SiO2(quartz) 0.00000 0.90635 SiO2(tridymite) 0.00000 0.99918 SiO2(cristobali) 1.00000 1.00000 SiO2(liquid) 0.00000 0.97125
/* Program cac14 */ /* Changing the Status of Phases */ #include "cacint.h" void table() { LI i, noerr, nphase; DB amount, act; char name[TQSTRLEN]; /* Retrieve and display the activities and equilibrium amounts of all phases */ /* Get number of phases */ tqnop(&nphase, &noerr); /* Print table header */ printf("%-27s%-16s%-16s\n", "Phase name", "amount/mol", "activity"); for(i = 1; i <= nphase; i++) { /* Get the phase name */ tqgnp(i, name, &noerr); /* Get its equilibrium amount */ tqgetr("a", i, 0, &amount, &noerr); /* Get its activity */ tqgetr("ac", i, 0, &act, &noerr); printf("%-24s%14.5f%14.5f\n", name, amount, act); } } int main() { LI noerr, numcon; LI iSiC, iSiO2, iC; DB vals[2]; char status[TQSTRLEN]; /* Initialise ChemApp */ tqini(&noerr); /* Open data-file for reading */ tqopna("cosi.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Get the phase index number of SiC */ tqinp("SiC", &iSiC, &noerr); /* Get the status of this phase */ tqgsp(iSiC, status, &noerr); printf("The status of SiC is currently %s\n", status);
The status of SiC is currently ENTERED
/* Unless a phase is marked "dormant" or "eliminated" in the data-file itself, the status is by default always "entered". This means it is considered in the subsequent equilibrium calculations and is included in the mass balance. */ /* Set some conditions and perform a simple calculation to demonstrate the effect of the different statuses */ /* Set temperature to 1900 K */ tqsetc("T", 0, 0, 1900.0, &numcon, &noerr); /* Set incoming amount to 1 mol SiO2(quartz) and 2 mol C */ tqinp("SiO2(quartz)", &iSiO2, &noerr); tqsetc("ia", iSiO2, 0, 1.0, &numcon, &noerr); tqinp("C", &iC, &noerr); tqsetc("ia", iC, 0, 2.0, &numcon, &noerr); /* Calculate the equilibrium */ tqce(" ", 0, 0, vals, &noerr); /* Print a table containing the phase names, their equilibrium amounts, and activities */ table();
Phase name amount/mol activity GAS 1.39778 1.00000 C 0.00000 0.43860 Si 0.00000 0.07912 SiC 0.65059 1.00000 SiO2(quartz) 0.00000 0.90635 SiO2(tridymite) 0.00000 0.99918 SiO2(cristobali) 0.30105 1.00000 SiO2(liquid) 0.00000 0.97125
/* From the above table it can be seen that SiC is stable under those conditions. Its activity is unity and its equilibrium amount is approx. 0.65 mol */ /* Now change the status of SiC to "dormant" and rerun the calculation */ tqcsp(iSiC, "dormant", &noerr); tqce(" ", 0, 0, vals, &noerr); table();
Phase name amount/mol activity GAS 0.00000 0.74210 C 2.00000 1.00000 Si 0.00000 1.00000 SiC 0.00000 28.81864 SiO2(quartz) 0.00000 0.90635 SiO2(tridymite) 0.00000 0.99918 SiO2(cristobali) 1.00000 1.00000 SiO2(liquid) 0.00000 0.97125
/* Setting a phase to "dormant" tells ChemApp to exclude it from the mass balance, but still calculate its activity. This offers the possibility to consider a phase to be metastable, while still obtaining information about its activity. It can be seen from the above table that ChemApp sets the equilibrium amount of SiC to zero, and calculated its activity to be almost 30. Instead of SiC, ChemApp now calculates Si to be stable. */ /* If a phase is to be completely ignored during the equilibrium calculations, its status should be set to "eliminated", as in the following example. */ tqcsp(iSiC, "eliminated", &noerr); tqce(" ", 0, 0, vals, &noerr); table(); /* Now ChemApp ignored SiC during the equilibrium calculations, as if it were not even included in the data-file. The status "eliminated" allows one to selectively delete phases from equilibrium calculations, for instance to increase computational efficiency. */ return 0; }
Phase name amount/mol activity GAS 0.00000 0.74210 C 2.00000 1.00000 Si 0.00000 1.00000 SiC 0.00000 0.00000 SiO2(quartz) 0.00000 0.90635 SiO2(tridymite) 0.00000 0.99918 SiO2(cristobali) 1.00000 1.00000 SiO2(liquid) 0.00000 0.97125
3.24 TQCSP
CHANGE-STATUS-OF-PHASE
Use TQCSP to change the status of a specified phase.
Synopsis
- FORTRAN:
- CALL TQCSP(INDEXP,OPTION,NOERR)
- C:
- tqcsp(indexp,option,&noerr);
- Pascal:
- tqcsp(indexp,option,noerr);
- Basic:
- Call tqcsp(indexp,option,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| OPTION | CHARACTER | Set to a string as shown in Table Possible statuses for phases and constituents |
| NOERR | INTEGER | Returns an error number |
INDEXP is integer input and OPTION is character input identifying the status (see Table Possible statuses for phases and constituents ).
- See
- TQGSP
3.25 TQGSPC
GET-STATUS-OF-PHASE-CONSTITUENT
Use TQGSPC to get the status of a specified phase constituent.
Synopsis
- FORTRAN:
- CALL TQGSPC(INDEXP,INDEXC,OPTION,NOERR)
- C:
- tqgspc(indexp,indexc,option,&noerr);
- Pascal:
- tqgspc(indexp,indexc,option,noerr);
- Basic:
- Call tqgspc(indexp,indexc,option,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXC | INTEGER | Set to the index number for a constituent |
| OPTION | CHARACTER | Returns a string as shown in Table Possible statuses for phases and constituents |
| NOERR | INTEGER | Returns an error number |
INDEXP and INDEXC are integer input and OPTION is character output identifying the status (see Table Possible statuses for phases and constituents).
! Changing the status of phase constituents SUBROUTINE TABLE IMPLICIT NONE INTEGER I, NOERR, NPCON DOUBLE PRECISION AMOUNT, FUG CHARACTER NAME*24, STATUS*24 ! Retrieve and display the fugacities, equilibrium ! amounts, and statuses of all phase constituents in the gas phase ! Get number of gas phase constituents CALL TQNOPC(1, NPCON, NOERR) ! Print table header WRITE(*,FMT='(A17,9X,A14,2X,A14,3X,A6)') * 'Phase const. name', * 'amount/mol', * 'fugacity', * 'status' DO I=1, NPCON ! Get the phase constituent name CALL TQGNPC(1, I, NAME, NOERR) ! Get its equilibrium amount CALL TQGETR('A ', 1, I, AMOUNT, NOERR) ! Get its fugacity CALL TQGETR('AC ', 1, I, FUG, NOERR) ! Get its status CALL TQGSPC(1, I, STATUS, NOERR) WRITE(*,FMT='(A24,2X,G14.5,2X,G14.5,2X,A10)') * NAME, AMOUNT, FUG, STATUS ENDDO RETURN END PROGRAM CAF15 IMPLICIT NONE INTEGER I, NOERR, NUMCON, NPCON INTEGER ISIO2, IC, ISI DOUBLE PRECISION VALS(2), STOI(3), WMASS ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Unless a phase constituent is marked "dormant" or "eliminated" in the ! data-file itself, the status is by default always "entered". This ! means it is considered in the subsequent equilibrium calculation and ! is included in the mass balances. ! Set some conditions and perform a simple calculation to ! demonstrate the effect of the different statuses ! Set temperature to 1900 K CALL TQSETC('T ', 0, 0, 1900.D0, NUMCON, NOERR) ! Set incoming amount to 1 mol SiO2(quartz) and 2 mol C CALL TQINP('SiO2(quartz)', ISIO2, NOERR) CALL TQSETC('IA ', ISIO2, 0, 1.D0, NUMCON, NOERR) CALL TQINP('C ', IC, NOERR) CALL TQSETC('IA ', IC, 0, 2.D0, NUMCON, NOERR) ! Calculate the equilibrium CALL TQCE(' ', 0, 0, VALS, NOERR) ! Print a table containing the phase constituent names, their ! equilibrium amounts, activities, and status CALL TABLE
Phase const. name amount/mol fugacity status C 0.19355E-11 0.13847E-11 ENTERED C2 0.42921E-13 0.30707E-13 ENTERED C3 0.29728E-12 0.21268E-12 ENTERED CO 1.3493 0.96531 ENTERED CO2 0.12171E-03 0.87076E-04 ENTERED O 0.21225E-10 0.15185E-10 ENTERED O2 0.35711E-14 0.25548E-14 ENTERED O3 0.60783E-29 0.43486E-29 ENTERED Si 0.13427E-05 0.96060E-06 ENTERED Si2 0.94642E-09 0.67709E-09 ENTERED Si2C 0.23037E-07 0.16481E-07 ENTERED Si3 0.16040E-10 0.11475E-10 ENTERED SiC 0.74337E-11 0.53182E-11 ENTERED SiO 0.48362E-01 0.34599E-01 ENTERED SiO2 0.55877E-07 0.39975E-07 ENTERED
! To demonstrate the effect of assigning a different status to phase ! constituents, set the status of all those phase constituents of the ! gas phase to "dormant" which contain Si. ! Get number of gas phase constituents CALL TQNOPC(1, NPCON, NOERR) ! Get the index number of system component Si CALL TQINSC("Si", ISI, NOERR) ! Use TQSTPC to determine for each phase constituent whether it ! contains Si, and set its status to "dormant" if it does DO I=1, NPCON CALL TQSTPC(1, I, STOI, WMASS, NOERR) IF (STOI(ISI) .GT. 0.D0) THEN CALL TQCSPC(1, I, 'DORMANT ', NOERR) ENDIF ENDDO ! Calculate the equilibrium and print the table again CALL TQCE(' ', 0, 0, VALS, NOERR) CALL TABLE ! As can be seen from the table above, all phase constituents of the gas ! phase that contain Si have been set to "dormant". ChemApp considers ! them in the activity calculation, but does not include them in the ! mass balances: their equilibrium amount is always zero. Since this ! forces ChemApp to keep the gas phase free of Si compounds, the 1 mol ! of Si input into the system can thus only be found in the condensed ! phases. ! Setting the status of these phase constituents to "eliminated" instead ! would cause ChemApp to exclude them completely from the equilibrium ! calculation. END
Phase const. name amount/mol fugacity status C 0.18901E-11 0.14176E-11 ENTERED C2 0.42909E-13 0.32183E-13 ENTERED C3 0.30425E-12 0.22820E-12 ENTERED CO 1.3332 0.99991 ENTERED CO2 0.12168E-03 0.91262E-04 ENTERED O 0.20485E-10 0.15364E-10 ENTERED O2 0.34873E-14 0.26155E-14 ENTERED O3 0.60057E-29 0.45044E-29 ENTERED Si 0.0000 0.93832E-06 DORMANT Si2 0.0000 0.64603E-09 DORMANT Si2C 0.0000 0.16099E-07 DORMANT Si3 0.0000 0.10695E-10 DORMANT SiC 0.0000 0.53182E-11 DORMANT SiO 0.0000 0.34195E-01 DORMANT SiO2 0.0000 0.39975E-07 DORMANT
/* Program cac15 */ /* Changing the status of phase constituents*/ #include "cacint.h" void table() { LI i, noerr, npcon; DB amount, fug; char name[TQSTRLEN], status[TQSTRLEN]; /* Retrieve and display the fugacities, equilibrium amounts, and statuses of all phase constituents in the gas phase */ /* Get number of phase constituents */ tqnopc(1, &npcon, &noerr); /* Print table header */ printf("%-24s %-12s %-12s%s\n", "Phase const. name", "amount/mol", "fugacity", "status"); for(i = 1; i <= npcon; i++) { /* Get the phase constituent name */ tqgnpc(1, i, name, &noerr); /* Get its equilibrium amount */ tqgetr("a", 1, i, &amount, &noerr); /* Get its fugacity */ tqgetr("ac", 1, i, &fug, &noerr); /* Get its status */ tqgspc(1, i, status, &noerr); printf("%-24s %10.5g %10.5g %-6s\n", name, amount, fug, status); } } int main() { LI i, noerr, numcon, npcon; LI iSiO2, iC, iSi; DB vals[2], stoi[3], wmass; /* Initialise ChemApp */ tqini(&noerr); /* Open data-file for reading */ tqopna("cosi.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Unless a phase constituent is marked "dormant" or "eliminated" in the data-file itself, the status is by default always "entered". This means it is considered in the subsequent equilibrium calculation and is included in the mass balances. */ /* Set some conditions and perform a simple calculation to demonstrate the effect of the different statuses */ /* Set temperature to 1900 K */ tqsetc("T", 0, 0, 1900.0, &numcon, &noerr); /* Set incoming amount to 1 mol SiO2(quartz) and 2 mol C */ tqinp("SiO2(quartz)", &iSiO2, &noerr); tqsetc("ia", iSiO2, 0, 1.0, &numcon, &noerr); tqinp("C", &iC, &noerr); tqsetc("ia", iC, 0, 2.0, &numcon, &noerr); /* Calculate the equilibrium */ tqce(" ", 0, 0, vals, &noerr); /* Print a table containing the phase constituent names, their equilibrium amounts, activities, and status */ table();
Phase const. name amount/mol fugacity status C 1.9355e-12 1.3847e-12 ENTERED C2 4.2921e-14 3.0707e-14 ENTERED C3 2.9728e-13 2.1268e-13 ENTERED CO 1.3493 0.96531 ENTERED CO2 0.00012171 8.7076e-05 ENTERED O 2.1225e-11 1.5185e-11 ENTERED O2 3.5711e-15 2.5548e-15 ENTERED O3 6.0783e-30 4.3486e-30 ENTERED Si 1.3427e-06 9.606e-07 ENTERED Si2 9.4642e-10 6.7709e-10 ENTERED Si2C 2.3037e-08 1.6481e-08 ENTERED Si3 1.604e-11 1.1475e-11 ENTERED SiC 7.4337e-12 5.3182e-12 ENTERED SiO 0.048362 0.034599 ENTERED SiO2 5.5877e-08 3.9975e-08 ENTERED
/* To demonstrate the effect of assigning a different status to phase constituents, set the status of all those phase constituents of the gas phase to "dormant" which contain Si. */ /* Get number of gas phase constituents */ tqnopc(1, &npcon, &noerr); /* Get the index number of system component Si */ tqinsc("Si", &iSi, &noerr); /* Use tqstpc to determine for each phase constituent whether it contains Si, and set its status to "dormant" if it does */ for (i = 1; i <= npcon; i++) { tqstpc(1, i, stoi, &wmass, &noerr); if (stoi[iSi-1] > 0.0) { tqcspc(1, i, "dormant", &noerr); } } /* Calculate the equilibrium and print the table again */ tqce(" ", 0, 0, vals, &noerr); table(); /* As can be seen from the table above, all phase constituents of the gas phase that contain Si have been set to "dormant". ChemApp considers them in the activity calculation, but does not include them in the mass balances: their equilibrium amount is always zero. Since this forces ChemApp to keep the gas phase free of Si compounds, the 1 mol of Si input into the system can thus only be found in the condensed phases. */ /* Setting the status of these phase constituents to "eliminated" instead would cause ChemApp to exclude them completely from the equilibrium calculation. */ return 0; }
Phase const. name amount/mol fugacity status C 1.8901e-12 1.4176e-12 ENTERED C2 4.2909e-14 3.2183e-14 ENTERED C3 3.0425e-13 2.282e-13 ENTERED CO 1.3332 0.99991 ENTERED CO2 0.00012168 9.1262e-05 ENTERED O 2.0485e-11 1.5364e-11 ENTERED O2 3.4873e-15 2.6155e-15 ENTERED O3 6.0057e-30 4.5044e-30 ENTERED Si 0 9.3832e-07 DORMANT Si2 0 6.4603e-10 DORMANT Si2C 0 1.6099e-08 DORMANT Si3 0 1.0695e-11 DORMANT SiC 0 5.3182e-12 DORMANT SiO 0 0.034195 DORMANT SiO2 0 3.9975e-08 DORMANT
3.26 TQCSPC
CHANGE-STATUS-OF-PHASE-CONSTITUENT
Use TQCSPC to change the status of a specified phase constituent.
Synopsis
- FORTRAN:
- CALL TQCSPC(INDEXP,INDEXC,OPTION,NOERR)
- C:
- tqcspc(indexp,indexc,option,&noerr);
- Pascal:
- tqcspc(indexp,indexc,option,noerr);
- Basic:
- Call tqcspc(indexp,indexc,option,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXC | INTEGER | Set to the index number for a constituent |
| OPTION | CHARACTER | Set to a string as shown in Table Possible statuses for phases and constituents |
| NOERR | INTEGER | Returns an error number |
INDEXP and INDEXC are integer input, and OPTION is character input identifying the status (see Table Possible statuses for phases and constituents).
Whereas complete phases can always be set ENTERED, DORMANT or ELIMINATED, some restrictions apply for phase constituents.
- For phases described by the models SUBS and QUAS, the status for single phase constituents cannot be changed.
- For sublattice type phases or for phases which internally are considered to contain sublattices (models SUBL, SUBO, SUBI, SUBM, SUBE, QUSL, GAYE, QSOL), the entered phase constituents must constitute a proper sublattice phase. As an example, if the sublattice setting is (A,B)(C,D), the phase constituents AC and AD, and thus implicitly A, can be removed, but not AC and BD. If there is an error, the error message appears first after a call of TQCE.
- See
- TQGSPC
4 Subroutines for Defining Equilibrium Calculations
This section describes the subroutines available for defining the initial conditions for an equilibrium calculation. These are summarised in Table ChemApp subroutines for defining calculations. Global equilibrium conditions are set using the subroutines TQSETC or TQSTEC (see Tables Possible state variables for setting conditions in the subroutines TQSETC and TQSTEC and Definition of the indices in the subroutines TQSETC and TQSTEC). The latter must be applied when using streams, which should preferably be defined when an extensive property of a reaction is wanted as a result, or when incoming amounts of the same constituent are to be entered more than once.
TQREMC is used to remove equilibrium conditions when using global conditions. All remaining subroutines in this group are used for managing streams. They set up a stream (TQSTTP), set its constituent amounts (TQSTCA), and delete a stream (TQSTRM).
| Subroutine | Function |
|---|---|
| TQSETC | Sets an equilibrium condition |
| TQREMC | Removes an equilibrium condition |
| TQSTTP | Sets name, temperature, and pressure for a stream |
| TQSTCA | Sets constituent amounts for a stream |
| TQSTEC | Sets an equilibrium condition with input conditions defined by streams |
| TQSTRM | Removes a stream |
| Option | Variable | Comment |
|---|---|---|
| P | Total pressure | Last entered (pressure or volume) takes precedence. Default value is 1 bar. |
| VT | Total volume | Last entered (pressure or volume) takes precedence. Note that the use of this option to perform calculations at constant volume is discouraged, see Equilibrium calculations at constant volume for details. |
| T | Temperature | Default value is 1000 K. |
| A | Amount of phase | VAL >=0 defines a formation target calculation; VAL <0 a precipitation target calculation for a mixture phase. In a formation target calculation, the target variable is varied until unit activity and zero amount of the target phase is achieved. A precipitation target is defined as one where the target phase is the only one formed at equilibrium, and the second most stable phase has unit activity. |
| IA | Incoming amount | Valid for phase constituents and system components. Not permitted for TQSTEC. |
| MU | Chemical potential | Valid only for phase constituents. Not permitted for TQSTEC. |
| AC | Relative activity | Not permitted for TQSTEC. |
CP
H
S
G
V
|
Heat capacity
Enthalpy
Entropy
Gibbs energy
Volume
|
Valid for the entire system or for a phase. Extensive properties are either calculated for the equilibrium phases and are then dependent on the reference state chosen in the data-file, or for the chemical reaction when streams are defined, in which case they are relative to the state defined by the stream conditions. |
| AT | Relative activity | Relative activity of a phase constituent as target. |
| XT | Mole fraction | Mole fraction of a phase constituent as target. |
| XPT | Mole fraction | Mole fraction of a system component in a phase as target. |
| INDEXP | INDEX | Component, phase, or constituent for which value is entered |
|---|---|---|
| >0 | >0 | Constituent INDEX of phase INDEXP (INDEX=1 or 0 for stoichiometric condensed phases) |
| >0 | <=0 | Phase INDEXP |
| <=0 | >0 | System component INDEX |
| 0 | 0 | Entire system |
4.1 TQSETC
SET-EQUILIBRIUM-CONDITION
Use TQSETC to set a condition for the equilibrium calculation.
Synopsis
- FORTRAN:
- CALL TQSETC(OPTION,INDEXP,INDEX,VAL,NUMCON,NOERR)
- C:
- tqsetc(option,indexp,index,val,&numcon,&noerr);
- Pascal:
- tqsetc(option,indexp,index,val,numcon,noerr);
- Basic:
- Call tqsetc(option,indexp,index,val,numcon,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table Possible state variables for setting conditions in the subroutines TQSETC and TQSTEC |
| INDEXP | INTEGER | Set to the index number for a phase, if necessary |
| INDEX | INTEGER | Set to the index number for a constituent or component, if necessary |
| VAL | DOUBLE PRECISION | Set to the value of the condition |
| NUMCON | INTEGER | Returns a number for the condition |
| NOERR | INTEGER | Returns an error number |
OPTION, INDEXP, INDEX, and VAL are all input. NUMCON is output. The possible state variables are shown in Table Possible state variables for setting conditions in the subroutines TQSETC and TQSTEC, and possible values of INDEXP and INDEX in Table Definition of the indices in the subroutines TQSETC and TQSTEC. VAL is entered with the default unit, or a unit set using TQCSU (see Initialising the Interface and Reading a Data-file). Each condition with respect to incoming amount, chemical potential, or relative activity is given a unique number (NUMCON). This number can later be used to remove any of these conditions (see TQREMC).
Normally two state variables, such as temperature and pressure, as well as one condition for the amount of each component of the actual system are necessary to define a calculation. The latter condition can also be fulfilled by defining incoming amounts of any number of constituents. Note that the input amounts entered do not have to include all the elements contained in the data-file. A previous value is replaced if incoming amounts for the same constituent are entered twice.
ChemApp defines a constituent for which chemical potential or relative activity is entered as incoming. The incoming amount (negative or positive) that corresponds to the condition set is calculated and the results can be retrieved using TQGETR. When any other variable than pressure (volume), temperature, or input composition is defined as an equilibrium condition, for example, enthalpy or the amount of a phase, a target variable has to be entered upon calling TQCE, see Subroutines to Perform a Calculation and to Obtain Results. Note that TQSETC and TQSTCA/TQSTEC cannot be used interchangeably for setting conditions.
Note that some solution phase models might not permit the usage of their phase constituents as incoming species. TQPCIS can be used to check for these cases.
! Set and remove equilibrium conditions PROGRAM CAF16 IMPLICIT NONE INTEGER NOERR, NUMCON, ISIO2, IC, ISIC, ISI DOUBLE PRECISION VALS(2), RESULT ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Set conditions for a simple equilibrium calculation: temperature and ! some incoming amounts. ! Set temperature to 1900 K CALL TQSETC('T ', 0, 0, 1900.D0, NUMCON, NOERR) ! Set incoming amounts: 1 mol SiO2(quartz), 2 mol C, and 3 mol SiC ! Get phase index numbers for all phases and set their amounts CALL TQINP('SiO2(quartz)', ISIO2, NOERR) CALL TQSETC('IA ', ISIO2, 1, 1.D0, NUMCON, NOERR) CALL TQINP('C ', IC, NOERR) CALL TQSETC('IA ', IC, 0, 2.D0, NUMCON, NOERR) CALL TQINP('SiC ', ISIC, NOERR) CALL TQSETC('IA ', ISIC, 0, 3.D0, NUMCON, NOERR) ! Use TQSHOW to check which conditions have been set so far, or are ! active by default CALL TQSHOW(NOERR)
T = 1900.00000000 K P = 1 bar STREAM CONSTITUENT SiO2(quartz) AMOUNT/mol = 1.00000000000000E+00 C AMOUNT/mol = 2.00000000000000E+00 SiC AMOUNT/mol = 3.00000000000000E+00 INACTIVE ELECTRON CONSTRAINT
! TQSETC returns a variable (called NUMCON in this example) which can be ! used to remove a previously set condition. To do this, pass it to ! TQREMC. Here we remove the last condition set, which are the 3 mol of ! SiC CALL TQREMC(NUMCON, NOERR) ! Call TQSHOW again to demonstrate that it has been deleted from the ! list of active conditions CALL TQSHOW(NOERR)
T = 1900.00000000 K P = 1 bar STREAM CONSTITUENT SiO2(quartz) AMOUNT/mol = 1.00000000000000E+00 C AMOUNT/mol = 2.00000000000000E+00 INACTIVE ELECTRON CONSTRAINT
! Calculate the equilibrium CALL TQCE(' ', 0, 0, VALS, NOERR) ! Get the activity of Si CALL TQINP('Si ', ISI, NOERR) CALL TQGETR('AC ', ISI, 0, RESULT, NOERR) WRITE(*,FMT='(1X,A,G12.5)') 'Activity of pure Si is ', RESULT END
Activity of pure Si is 0.79115E-01
/* Program cac16 */ /* Set equilibrium conditions */ #include "cacint.h" int main() { LI noerr, numcon, iSiO2, iC, iSiC, iSi; DB vals[2], result; /* Initialise ChemApp */ tqini(&noerr); /* Open data-file for reading */ tqopna("cosi.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Set conditions for a simple equilibrium calculation: temperature and some incoming amounts. */ /* Set temperature to 1900 K */ tqsetc("T", 0, 0, 1900.0, &numcon, &noerr); /* Set incoming amounts: 1 mol SiO2(quartz), 2 mol C, and 3 mol SiC */ /* Get phase index number for both phases and set their amounts */ tqinp("SiO2(quartz)", &iSiO2, &noerr); tqsetc("ia", iSiO2, 0, 1.0, &numcon, &noerr); tqinp("C", &iC, &noerr); tqsetc("ia", iC, 0, 2.0, &numcon, &noerr); tqinp("SiC", &iSiC, &noerr); tqsetc("ia", iSiC, 0, 3.0, &numcon, &noerr); /* Use tqshow to check which conditions have been set so far, or are active by default */ tqshow(&noerr);
T = 1900.00000000 K P = 1 bar STREAM CONSTITUENT SiO2(quartz) AMOUNT/mol = 1.00000000000000E+00 C AMOUNT/mol = 2.00000000000000E+00 SiC AMOUNT/mol = 3.00000000000000E+00 INACTIVE ELECTRON CONSTRAINT
/* tqsetc returns a variable (called numcon in this example) which can be used to remove a previously set condition. To do this, pass it to tqremc. Here we remove the last condition set, which are the 3 mol of SiC */ tqremc(numcon, &noerr); /* Call tqshow again to demonstrate that it has been deleted from the list of active conditions */ tqshow(&noerr);
T = 1900.00000000 K P = 1 bar STREAM CONSTITUENT SiO2(quartz) AMOUNT/mol = 1.00000000000000E+00 C AMOUNT/mol = 2.00000000000000E+00 INACTIVE ELECTRON CONSTRAINT
/* Calculate the equilibrium */ tqce(" ", 0, 0, vals, &noerr); /* Get the activity of Si */ tqinp("Si", &iSi, &noerr); tqgetr("ac", iSi, 0, &result, &noerr); printf("Activity of pure Si is %g\n", result); return 0; }
Activity of pure Si is 0.0791154
4.2 TQREMC
REMOVE-EQUILIBRIUM-CONDITION
Use TQREMC to remove a condition for the equilibrium calculation.
Synopsis
- FORTRAN:
- CALL TQREMC(NUMCON,NOERR)
- C:
- tqremc(numcon,&noerr);
- Pascal:
- tqremc(numcon,noerr);
- Basic:
- Call tqremc(numcon,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NUMCON | INTEGER | Set the number for the condition |
| NOERR | INTEGER | Returns an error number |
Replace NUMCON with the name of the variable that was used for setting the condition (TQSETC) to be removed. TQREMC is needed only for the case when a condition with respect to an incoming amount, chemical potential, or relative activity (see Table Possible state variables for setting conditions in the subroutines TQSETC and TQSTEC) is to be removed. Otherwise, input conditions can be replaced by using another call to TQSETC, TQCE, or TQMAP (see Subroutines to Perform a Calculation and to Obtain Results).
Three special cases for the removal of conditions are provided, which are also accessed through the value of NUMCON passed to TQREMC:
| 0: | Using a zero value for NUMCON removes all input conditions relating to incoming amounts, but no other conditions (e.g. temperature), and no targets. |
|---|---|
| -1: | A value of -1 removes all conditions, targets, and previously set configuration options, plus it resets the system units to their default values. |
| -2: | A value of -2 does the same as the above, except that it does not reset the system units to their default values (new as of version 3.0.0). |
- See
- TQSETC
4.3 TQSTTP
SET-NAME-TEMPERATURE-PRESSURE-FOR-A-STREAM
Use TQSTTP to define a stream and set its temperature and pressure.
Synopsis
- FORTRAN:
- CALL TQSTTP(IDENTS,VALS,NOERR)
- C:
- tqsttp(idents,vals,&noerr);
- Pascal:
- tqsttp(idents,vals,noerr);
- Basic:
- Call tqsttp(idents,vals,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| IDENTS | CHARACTER | Set to a name for the stream |
| VALS | Array of DOUBLE PRECISION | Set to temperature and pressure for a stream |
| NOERR | INTEGER | Returns an error number |
The input variable IDENTS can have a maximum length of 150 characters. VALS must be an array of 2 real values, containing the temperature and pressure of the stream. These are entered with the default unit, or a unit set using TQCSU (see Initialising the Interface and Reading a Data-file).
A stream is a means for transferring non-reacted matter (see also the notes in Extensive properties of streams) to a reaction zone. It has constant temperature and pressure, and contains one or more phases of fixed composition. (see also Setting initial conditions). Stream input is required whenever differences in extensive properties between the initial and the equilibrium state are desired; for example, in calculations of the heat evolving from a combustion process, or of the entropy change caused by a phase transition.
The maximum number of streams which are defined must not be greater than the maximum number of constituents permitted in the ChemApp version currently used (see TQSIZE, parameter 'NA').
! Equilibrium calculations using streams PROGRAM CAF17 IMPLICIT NONE INTEGER NOERR, ISIO2, IO2, ICO2, I, NPHASE DOUBLE PRECISION TPSOL(2), TPGAS(2), VALS(2), AMOUNT, * VALUE CHARACTER PNAME*24, SNAME*24, CEU*3, CTU*1, CVU*3, CAU*5 ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Two input streams will subsequently be defined: The first one, ! labelled 'STREAM_QUARTZ', will be solid SiO2(quartz) at 500 K, the ! second one, labelled 'STREAM_GAS', will consist of a gas phase at room ! temperature consisting of O2 and CO2. By using streams instead of ! global conditions we will be able to get information about the ! extensive property balance of this process. ! Define the first stream ('STREAM_QUARTZ') ! Use the array called TPSOL to pass the information on temperature and ! pressure (500 K and 1 bar) of the stream to TQSTTP TPSOL(1) = 500.D0 TPSOL(2) = 1.D0 CALL TQSTTP('STREAM_QUARTZ ', TPSOL, NOERR) ! Define the amounts of constituents of this stream (1 mol quartz) CALL TQINP('SiO2(quartz)', ISIO2, NOERR) CALL TQSTCA('STREAM_QUARTZ ', ISIO2, 0, 1.D0, NOERR) ! Define second stream ('STREAM_GAS') TPGAS(1) = 298.15D0 TPGAS(2) = 1.D0 CALL TQSTTP('STREAM_GAS ', TPGAS, NOERR) ! Define the amounts of constituents of this stream (0.5 mol O2/GAS/ and ! 0.5 mol CO2/GAS/) CALL TQINPC('O2 ', 1, IO2, NOERR) CALL TQSTCA('STREAM_GAS ', 1, IO2, 0.5D0, NOERR) CALL TQINPC('CO2 ', 1, ICO2, NOERR) CALL TQSTCA('STREAM_GAS ', 1, ICO2, 0.5D0, NOERR) ! Set the temperature at which the two streams should react CALL TQSTEC('T ', 0, 2500.D0, NOERR) ! Define a third stream for the sole purpose of demonstrating how to ! remove it subsequently CALL TQSTTP('DUMMY ', TPGAS, NOERR) CALL TQSTCA('DUMMY ', 1, 1, 1.D0, NOERR) ! Display what we have set so far CALL TQSHOW(NOERR)
T = 2500.00000000 K
P = 1 bar
STREAM NAME STREAM NUMBER TEMPERATURE/K PRESSURE/bar
STREAM_QUARTZ
1 500.00 1.0000E+00
STREAM_GAS
2 298.15 1.0000E+00
DUMMY
3 298.15 1.0000E+00
STREAM NUMBER STREAM CONSTITUENT
1 SiO2(quartz)
AMOUNT/mol = 1.00000000000000E+00
2 O2/GAS/
AMOUNT/mol = 5.00000000000000E-01
2 CO2/GAS/
AMOUNT/mol = 5.00000000000000E-01
3 C/GAS/
AMOUNT/mol = 1.00000000000000E+00
INACTIVE ELECTRON CONSTRAINT
! Remove the 'DUMMY' stream CALL TQSTRM('DUMMY ', NOERR) ! Call TQSHOW again to demonstrate that the last stream has been removed CALL TQSHOW(NOERR)
T = 2500.00000000 K
P = 1 bar
STREAM NAME STREAM NUMBER TEMPERATURE/K PRESSURE/bar
STREAM_QUARTZ
1 500.00 1.0000E+00
STREAM_GAS
2 298.15 1.0000E+00
STREAM NUMBER STREAM CONSTITUENT
1 SiO2(quartz)
AMOUNT/mol = 1.00000000000000E+00
2 O2/GAS/
AMOUNT/mol = 5.00000000000000E-01
2 CO2/GAS/
AMOUNT/mol = 5.00000000000000E-01
INACTIVE ELECTRON CONSTRAINT
! Calculate the equilibrium CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 2500 K
P = 1 bar
V = 211.73 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/K PRESSURE/bar STREAM
SiO2(quartz) 1.0000E+00 500.00 1.0000E+00 1
O2/GAS/ 5.0000E-01 298.15 1.0000E+00 2
CO2/GAS/ 5.0000E-01 298.15 1.0000E+00 2
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
O2 5.0755E-01 4.9829E-01 4.9829E-01
CO2 4.7466E-01 4.6599E-01 4.6599E-01
CO 2.5340E-02 2.4877E-02 2.4877E-02
O 1.0519E-02 1.0327E-02 1.0327E-02
SiO 2.8827E-04 2.8300E-04 2.8300E-04
SiO2 2.3878E-04 2.3442E-04 2.3442E-04
O3 T 1.1157E-07 1.0954E-07 1.0954E-07
Si 2.7197E-12 2.6701E-12 2.6701E-12
C 8.5771E-16 8.4205E-16 8.4205E-16
Si2 4.6313E-23 4.5467E-23 4.5467E-23
SiC 1.0075E-23 9.8915E-24 9.8915E-24
C2 1.1127E-24 1.0924E-24 1.0924E-24
Si2C 4.9282E-30 4.8382E-30 4.8382E-30
C3 4.4137E-32 4.3332E-32 4.3332E-32
Si3 4.3927E-33 4.3125E-33 4.3125E-33
TOTAL: 1.0186E+00 1.0000E+00 1.0000E+00
mol ACTIVITY
SiO2(liquid) 9.9947E-01 1.0000E+00
SiO2(cristobali) 0.0000E+00 8.9012E-01
SiO2(tridymite) T 0.0000E+00 8.5795E-01
SiO2(quartz) T 0.0000E+00 7.4840E-01
C 0.0000E+00 5.0218E-09
Si 0.0000E+00 5.7669E-10
SiC 0.0000E+00 1.3051E-17
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
9.94401E+01 2.72660E+05 2.18581E+02 -8.84766E+05 1.86939E+02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.92216E+02 -8.24091E+05 5.02347E+02 -2.07996E+06 2.11729E+02
Mole fraction of system components:
GAS
C 0.19987
O 0.79992
Si 2.1068E-04
Data on 3 constituents marked with 'T' are extrapolated outside their valid
temperature range
50 FORMAT(1X,A,G12.5,' ',A) ! Get the current system units CALL TQGSU('V ', CVU, NOERR) CALL TQGSU('T ', CTU, NOERR) CALL TQGSU('E ', CEU, NOERR) CALL TQGSU('A ', CAU, NOERR) ! Get the list of stable phases WRITE(*,FMT='(A)') ' List of stable phases:' CALL TQNOP(NPHASE, NOERR) DO I=1, NPHASE CALL TQGETR('A ', I, 0, AMOUNT, NOERR) IF (AMOUNT .GT. 0) THEN CALL TQGNP(I, PNAME, NOERR) WRITE(*,FMT=50) 'Phase: ' // PNAME // * 'amount: ', AMOUNT, CAU ENDIF ENDDO
List of stable phases: Phase: GAS amount: 1.0186 mol Phase: SiO2(liquid) amount: 0.99947 mol
! Get the changes in the extensive property values in the current units WRITE(*,'(A)') ' Changes in extensive properties:' CALL TQGETR('CP ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Delta CP: ', VALUE, CEU // '/' // CTU CALL TQGETR('H ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Delta H: ', VALUE, CEU CALL TQGETR('S ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Delta S: ', VALUE, CEU // '/' // CTU CALL TQGETR('G ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Delta G: ', VALUE, CEU CALL TQGETR('V ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Delta V: ', VALUE, CVU
Changes in extensive properties: Delta CP: 99.440 J /K Delta H: 0.27266E+06 J Delta S: 218.58 J /K Delta G: -0.88477E+06 J Delta V: 186.94 dm3
! To retrieve the *total* volume, use the option 'VT' CALL TQGETR('VT ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Total volume: ', VALUE, CVU
Total volume: 211.73 dm3
! Get the thermodynamic properties of the two streams (note that as ! of version 3.2.0 of ChemApp, TQSTXP can also be called _before_ ! the equilibrium calculation) SNAME = 'STREAM_GAS' CALL TQSTXP(SNAME, 'CP ', VALUE, NOERR) WRITE(*,FMT=50) 'Cp of stream '// SNAME // ': ', * VALUE, CEU // '/' // CTU CALL TQSTXP(SNAME, 'H ', VALUE, NOERR) WRITE(*,FMT=50) 'H of stream '// SNAME // ': ', * VALUE, CEU CALL TQSTXP(SNAME, 'S ', VALUE, NOERR) WRITE(*,FMT=50) 'S of stream '// SNAME // ': ', * VALUE, CEU // '/' // CTU CALL TQSTXP(SNAME, 'G ', VALUE, NOERR) WRITE(*,FMT=50) 'G of stream '// SNAME // ': ', * VALUE, CEU CALL TQSTXP(SNAME, 'V ', VALUE, NOERR) WRITE(*,FMT=50) 'V of stream '// SNAME // ': ', * VALUE, CVU
Cp of stream STREAM_GAS : 33.278 J /K H of stream STREAM_GAS : -0.19675E+06 J S of stream STREAM_GAS : 215.22 J /K G of stream STREAM_GAS : -0.26092E+06 J V of stream STREAM_GAS : 24.790 dm3
! Note that the volume of condensed phases is zero, unless ! their thermodynamic data contain molar volumes: SNAME = 'STREAM_QUARTZ' CALL TQSTXP(SNAME, 'V ', VALUE, NOERR) WRITE(*,FMT=50) 'Volume of stream '// SNAME // ': ', * VALUE, CVU
Volume of stream STREAM_QUARTZ : 0.0000 dm3
! Change the system units to show that TQSTXP returns values ! in the current system units CALL TQCSU('T ', 'F ', NOERR) CALL TQCSU('V ', 'FT3 ', NOERR) CALL TQCSU('E ', 'CAL ', NOERR) CALL TQGSU('V ', CVU, noerr) CALL TQGSU('T ', CTU, noerr) CALL TQGSU('E ', CEU, noerr) SNAME = 'STREAM_GAS' CALL TQSTXP(SNAME, 'CP ', VALUE, NOERR) WRITE(*,FMT=50) 'Cp of stream '// SNAME // ': ', * VALUE, CEU // '/' // CTU CALL TQSTXP(SNAME, 'H ', VALUE, NOERR) WRITE(*,FMT=50) 'H of stream '// SNAME // ': ', * VALUE, CEU CALL TQSTXP(SNAME, 'S ', VALUE, NOERR) WRITE(*,FMT=50) 'S of stream '// SNAME // ': ', * VALUE, CEU // '/' // CTU CALL TQSTXP(SNAME, 'G ', VALUE, NOERR) WRITE(*,FMT=50) 'G of stream '// SNAME // ': ', * VALUE, CEU CALL TQSTXP(SNAME, 'V ', VALUE, NOERR) WRITE(*,FMT=50) 'V of stream '// SNAME // ': ', * VALUE, CVU END
Cp of stream STREAM_GAS : 4.4187 cal/F H of stream STREAM_GAS : -47025. cal S of stream STREAM_GAS : 28.577 cal/F G of stream STREAM_GAS : -62362. cal V of stream STREAM_GAS : 0.87544 ft3
/* Program cac17 */ /* Equilibrium calculations using streams */ #include "cacint.h" int main() { LI noerr, iSiO2, iO2, iCO2, i, nphase; DB TP_sol[2], TP_gas[2], vals[2], amount, value; char pname[TQSTRLEN], sname[TQSTRLEN]; char CEU[TQSTRLEN], CTU[TQSTRLEN], CVU[TQSTRLEN], CAU[TQSTRLEN]; /* Initialise ChemApp */ tqini(&noerr); /* Open data-file for reading */ tqopna("cosi.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Two input streams will subsequently be defined: The first one, labelled "STREAM_QUARTZ", will be solid SiO2(quartz) at 500 K, the second one, labelled "STREAM_GAS", will consist of a gas phase at room temperature consisting of O2 and CO2. By using streams instead of global conditions we will be able to get information about the extensive property balance of this process. */ /* Define the first stream ("STREAM_QUARTZ") */ /* Use the array called TP_sol to pass the information on temperature and pressure (500 K and 1 bar) of the stream to tqsttp */ TP_sol[0] = 500.0; TP_sol[1] = 1.0; tqsttp("STREAM_QUARTZ", TP_sol, &noerr); /* Define the amounts of constituents of this stream (1 mol quartz)*/ tqinp("SiO2(quartz)", &iSiO2, &noerr); tqstca("STREAM_QUARTZ", iSiO2, 0, 1.0, &noerr); /* Define second stream ("STREAM_GAS") */ TP_gas[0] = 298.15; TP_gas[1] = 1.0; tqsttp("STREAM_GAS", TP_gas, &noerr); /* Define the amounts of constituents of this stream (0.5 mol O2/GAS/ and 0.5 mol CO2/GAS/) */ tqinpc("O2", 1, &iO2, &noerr); tqstca("STREAM_GAS", 1, iO2, 0.5, &noerr); tqinpc("CO2", 1, &iCO2, &noerr); tqstca("STREAM_GAS", 1, iCO2, 0.5, &noerr); /* Set the temperature at which the two streams should react */ tqstec("T", 0, 2500.0, &noerr); /* Define a third stream for the sole purpose of demonstrating how to remove it subsequently */ tqsttp("DUMMY", TP_gas, &noerr); tqstca("DUMMY", 1, 1, 1.0, &noerr); /* Display what we have set so far */ tqshow(&noerr);
T = 2500.00000000 K
P = 1 bar
STREAM NAME STREAM NUMBER TEMPERATURE/K PRESSURE/bar
STREAM_QUARTZ
1 500.00 1.0000E+00
STREAM_GAS
2 298.15 1.0000E+00
DUMMY
3 298.15 1.0000E+00
STREAM NUMBER STREAM CONSTITUENT
1 SiO2(quartz)
AMOUNT/mol = 1.00000000000000E+00
2 O2/GAS/
AMOUNT/mol = 5.00000000000000E-01
2 CO2/GAS/
AMOUNT/mol = 5.00000000000000E-01
3 C/GAS/
AMOUNT/mol = 1.00000000000000E+00
INACTIVE ELECTRON CONSTRAINT
/* Remove the "DUMMY" stream */ tqstrm("DUMMY", &noerr); /* Call tqshow again to demonstrate that the last stream has been removed */ tqshow(&noerr) ;
T = 2500.00000000 K
P = 1 bar
STREAM NAME STREAM NUMBER TEMPERATURE/K PRESSURE/bar
STREAM_QUARTZ
1 500.00 1.0000E+00
STREAM_GAS
2 298.15 1.0000E+00
STREAM NUMBER STREAM CONSTITUENT
1 SiO2(quartz)
AMOUNT/mol = 1.00000000000000E+00
2 O2/GAS/
AMOUNT/mol = 5.00000000000000E-01
2 CO2/GAS/
AMOUNT/mol = 5.00000000000000E-01
INACTIVE ELECTRON CONSTRAINT
/* Calculate the equilibrium */ tqcel(" ", 0, 0, vals, &noerr);
T = 2500 K
P = 1 bar
V = 211.73 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/K PRESSURE/bar STREAM
SiO2(quartz) 1.0000E+00 500.00 1.0000E+00 1
O2/GAS/ 5.0000E-01 298.15 1.0000E+00 2
CO2/GAS/ 5.0000E-01 298.15 1.0000E+00 2
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
O2 5.0755E-01 4.9829E-01 4.9829E-01
CO2 4.7466E-01 4.6599E-01 4.6599E-01
CO 2.5340E-02 2.4877E-02 2.4877E-02
O 1.0519E-02 1.0327E-02 1.0327E-02
SiO 2.8827E-04 2.8300E-04 2.8300E-04
SiO2 2.3878E-04 2.3442E-04 2.3442E-04
O3 T 1.1157E-07 1.0954E-07 1.0954E-07
Si 2.7197E-12 2.6701E-12 2.6701E-12
C 8.5771E-16 8.4205E-16 8.4205E-16
Si2 4.6313E-23 4.5467E-23 4.5467E-23
SiC 1.0075E-23 9.8915E-24 9.8915E-24
C2 1.1127E-24 1.0924E-24 1.0924E-24
Si2C 4.9282E-30 4.8382E-30 4.8382E-30
C3 4.4137E-32 4.3332E-32 4.3332E-32
Si3 4.3927E-33 4.3125E-33 4.3125E-33
TOTAL: 1.0186E+00 1.0000E+00 1.0000E+00
mol ACTIVITY
SiO2(liquid) 9.9947E-01 1.0000E+00
SiO2(cristobali) 0.0000E+00 8.9012E-01
SiO2(tridymite) T 0.0000E+00 8.5795E-01
SiO2(quartz) T 0.0000E+00 7.4840E-01
C 0.0000E+00 5.0218E-09
Si 0.0000E+00 5.7669E-10
SiC 0.0000E+00 1.3051E-17
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
9.94401E+01 2.72660E+05 2.18581E+02 -8.84766E+05 1.86939E+02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.92216E+02 -8.24091E+05 5.02347E+02 -2.07996E+06 2.11729E+02
Mole fraction of system components:
GAS
C 0.19987
O 0.79992
Si 2.1068E-04
Data on 3 constituents marked with 'T' are extrapolated outside their valid
temperature range
/* Get the current system units */ tqgsu("V", CVU, &noerr); tqgsu("T", CTU, &noerr); tqgsu("E", CEU, &noerr); tqgsu("A", CAU, &noerr); /* Get the list of stable phases */ printf("List of stable phases:\n"); tqnop(&nphase, &noerr); for (i = 1; i <= nphase; i++) { tqgetr("A", i, 0, &amount, &noerr); if (amount > 0.0) { tqgnp(i, pname, &noerr); printf("Phase: %-24s amount: %g %s\n", pname, amount, CAU); } }
List of stable phases: Phase: GAS amount: 1.0186 mol Phase: SiO2(liquid) amount: 0.999473 mol
/* Get the changes in the extensive property values in the current units */ printf("Changes in extensive properties:\n"); tqgetr("CP", 0, 0, &value, &noerr); printf("Delta CP: %g %s/%s\n", value, CEU, CTU); tqgetr("H", 0, 0, &value, &noerr); printf("Delta H : %g %s\n", value, CEU); tqgetr("S", 0, 0, &value, &noerr); printf("Delta S : %g %s/%s\n", value, CEU, CTU); tqgetr("G", 0, 0, &value, &noerr); printf("Delta G : %g %s\n", value, CEU); tqgetr("V", 0, 0, &value, &noerr); printf("Delta V : %g %s\n", value, CVU);
Changes in extensive properties: Delta CP: 99.4401 J/K Delta H : 272660 J Delta S : 218.581 J/K Delta G : -884766 J Delta V : 186.939 dm3
/* To retrieve the *total* volume, use the option "VT" */ tqgetr("VT", 0, 0, &value, &noerr); printf("Total volume: %g %s\n", value, CVU);
Total volume: 211.729 dm3
/* Get the thermodynamic properties of the two streams (note that as of version 3.2.0 of ChemApp, tqstxp can also be called _before_ the equilibrium calculation) */ strcpy(sname,"STREAM_GAS"); tqstxp(sname, "CP", &value, &noerr); printf("Cp of stream %-24s: %g %s/%s\n", sname, value, CEU, CTU); tqstxp(sname, "H", &value, &noerr); printf("H of stream %-24s: %g %s\n", sname, value, CEU); tqstxp(sname, "S", &value, &noerr); printf("S of stream %-24s: %g %s/%s\n", sname, value, CEU, CTU); tqstxp(sname, "G", &value, &noerr); printf("G of stream %-24s: %g %s\n", sname, value, CEU); tqstxp(sname, "V", &value, &noerr); printf("V of stream %-24s: %g %s\n", sname, value, CVU);
Cp of stream STREAM_GAS : 33.278 J/K H of stream STREAM_GAS : -196752 J S of stream STREAM_GAS : 215.221 J/K G of stream STREAM_GAS : -260921 J V of stream STREAM_GAS : 24.7897 dm3
/* Note that the volume of condensed phases is zero, unless their thermodynamic data contain molar volumes: */ strcpy(sname,"STREAM_QUARTZ"); tqstxp(sname, "V", &value, &noerr); printf("Volume of stream %-24s: %g %s\n", sname, value, CVU);
Volume of stream STREAM_QUARTZ : 0 dm3
/* Change the system units to show that tqstxp returns values in the current system units */ tqcsu("T", "F", &noerr); tqcsu("V", "FT3", &noerr); tqcsu("E", "CAL", &noerr); tqgsu("T", CTU, &noerr); tqgsu("V", CVU, &noerr); tqgsu("E", CEU, &noerr); strcpy(sname,"STREAM_GAS"); tqstxp(sname, "CP", &value, &noerr); printf("Cp of stream %-24s: %g %s/%s\n", sname, value, CEU, CTU); tqstxp(sname, "H", &value, &noerr); printf("H of stream %-24s: %g %s\n", sname, value, CEU); tqstxp(sname, "S", &value, &noerr); printf("S of stream %-24s: %g %s/%s\n", sname, value, CEU, CTU); tqstxp(sname, "G", &value, &noerr); printf("G of stream %-24s: %g %s\n", sname, value, CEU); tqstxp(sname, "V", &value, &noerr); printf("V of stream %-24s: %g %s\n", sname, value, CVU); return 0; }
Cp of stream STREAM_GAS : 4.41869 cal/F H of stream STREAM_GAS : -47025 cal S of stream STREAM_GAS : 28.5773 cal/F G of stream STREAM_GAS : -62361.5 cal V of stream STREAM_GAS : 0.875441 ft3
4.4 TQSTCA
SET-CONSTITUENT-AMOUNTS-FOR-A-STREAM
Use TQSTCA to set the constituent amounts for a stream.
Synopsis
- FORTRAN:
- CALL TQSTCA(IDENTS,INDEXP,INDEXC,VAL,NOERR)
- C:
- tqstca(idents,indexp,indexc,val,&noerr);
- Pascal:
- tqstca(idents,indexp,indexc,val,noerr);
- Basic:
- Call tqstca(idents,indexp,indexc,val,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| IDENTS | CHARACTER | Set to the name of a stream |
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXC | INTEGER | Set to the index number for a constituent |
| VAL | DOUBLE PRECISION | Set to a stream constituent amount |
| NOERR | INTEGER | Returns an error number |
IDENTS, INDEXP, INDEXC, and VAL are all input. IDENTS is a name previously defined by calling TQSTTP. VAL must be greater than zero and is entered with the default unit, or a unit set using TQCSU (see Initialising the Interface and Reading a Data-file).
See Extensive properties of streams for details on how ChemApp deals with the extensive properties of streams. Note that the same phase constituent can be a part of several streams.
The total number of all the stream constituents defined in all streams must not be greater than the maximum number of constituents of the version of ChemApp used (see TQSIZE, parameter 'NA'). This implies that this limit is reached when defining one stream with 'NA' constituents, or 'NA' streams with one constituent each.
Note that it is not possible to use TQSETC and TQSTCA/TQSTEC interchangeably for setting conditions.
Note that some solution phase models might not permit the usage of their phase constituents as incoming species. TQPCIS can be used to check for these cases.
- See
- TQSTTP
4.5 TQSTEC
SET-EQUILIBRIUM-CONDITION-WHEN-STREAM-INPUT
Use TQSTEC to set an invariant state variable for the equilibrium calculation when the input conditions are defined by streams.
Synopsis
- FORTRAN:
- CALL TQSTEC(OPTION,INDEXP,VAL,NOERR)
- C:
- tqstec(option,indexp,val,&noerr);
- Pascal:
- tqstec(option,indexp,val,noerr);
- Basic:
- Call tqstec(option,indexp,val,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table Possible state variables for setting conditions in the subroutines TQSETC and TQSTEC |
| INDEXP | INTEGER | Set to the index number for a phase, if necessary |
| VAL | DOUBLE PRECISION | Set to the value of the condition |
| NOERR | INTEGER | Returns an error number |
OPTION, INDEXP, and VAL are all input. See Table Possible state variables for setting conditions in the subroutines TQSETC and TQSTEC for the mnemonics of the possible state variables. VAL is entered with a default unit, or a unit set using TQCSU (see Chapter Initialising the Interface and Reading a Data-file).
Note: It is not possible to use TQSETC and TQSTCA/TQSTEC interchangeably for setting conditions.
- See
- TQSTTP
4.6 TQSTRM
REMOVE-STREAM
Use TQSTRM to remove a stream.
Synopsis
- FORTRAN:
- CALL TQSTRM(IDENTS,NOERR)
- C:
- tqstrm(idents,&noerr);
- Pascal:
- tqstrm(idents,noerr);
- Basic:
- Call tqstrm(idents,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| IDENTS | CHARACTER | Set to the name of a stream |
| NOERR | INTEGER | Returns an error number |
IDENTS is a name previously defined by calling TQSTTP.
The maximum number of streams that can be defined is equal to the number of constituents ChemApp can handle (see TQSIZE, parameter 'NA'). In the current version of ChemApp though, removing a previously defined stream does not free the number ChemApp used internally for this stream, i.e. the number of additional streams that can be defined is not increased by one. To remove all streams, assign an empty string (in FORTRAN a single blank) to IDENTS when calling TQSTRM. In this case, ChemApp internally deletes all streams and the next stream defined will be assigned to #1.
- See
- TQSTTP
5 Subroutines to Perform a Calculation and to Obtain Results
TQCE and TQCEL are the only subroutines that need to be called to carry out a single equilibrium calculation (TQCEL additionally displays results from a calculation in form of a ChemSage/ChemApp result table). When an equilibrium state variable other than pressure, temperature, or composition is defined as input, a target variable must be defined upon calling TQCE/TQCEL; for example, when defining enthalpy or the amount of a phase as a condition instead of temperature.
As of version 3.0.0, ChemApp is able to perform one-dimensional phase mapping calculations. This type of calculation is done using the subroutines TQMAP/TQMAPL.
TQSHOW displays a table of the present settings. Using TQCLIM it is possible to modify the upper and lower limits of target variables.
Results from an equilibrium calculation are retrieved using the subroutine TQGETR. Thermodynamic data for phase constituents can be obtained using TQGDPC, while thermodynamic properties of streams are accessible through TQSTXP. TQGTLC is used to get calculated equilibrium sublattice site fractions for phases modelled according to the sublattice formalism.
TQERR can be used to retrieve ChemApp error messages after any ChemApp subroutine indicated that an error occurred.
These subroutines are summarised below in Table ChemApp subroutines for calculating and getting results, while Tables State variables for defining targets when calling TQCE/TQCEL to State variables for defining one-dimensional phase mapping calculations using TQMAP/TQMAPL list possible values of variables used as parameters to the various subroutines in this group.
| Subroutine | Function |
|---|---|
| TQCE | Calculates the chemical equilibrium |
| TQCEL | Calculates the chemical equilibrium and gives a result table (ChemSage format) |
| TQCEN | Calculates the chemical equilibrium, taking results from the previous equilibrium calculation as initial estimates |
| TQCENL | Calculates the chemical equilibrium, taking results from the previous equilibrium calculation as initial estimates, and gives a result tab*e (ChemSage format) |
| TQMAP | Calculates a one-dimensional phase map |
| TQMAPL | Calculates a one-dimensional phase map and gives a result table (ChemSage format) |
| TQCLIM | Changes limits of target variables |
| TQSHOW | Shows present settings |
| TQGETR | Gets calculated equilibrium results |
| TQGDPC | Gets thermodynamic data for a phase constituent |
| TQSTXP | Gets thermodynamic properties of a stream |
| TQGTLC | Gets the calculated equilibrium sublattice site fraction |
| TQBOND | Gets a calculated quadruplet or pair fraction |
| TQERR | Gets an error message |
| Option | Variable | Comment |
|---|---|---|
| P | Total pressure | Pressure is a target variable. The value VALS(1) serves as an initial estimate, VALS(2) is not used. |
| V | Total volume | Volume is a target variable. The value VALS(1) serves as an initial estimate, VALS(2) is not used. |
| T | Temperature | Temperature is a target variable. The value VALS(1) serves as an initial estimate, VALS(2) is not used. |
IA
IA0
MU
|
Incoming amount
Chemical potential
|
Incoming amount is a target variable. The values VALS(1) and VALS(2) denote lower and upper limits, respectively. When defining streams, the constituent is included in the last stream considered. If incoming amounts for more then one substance need to be defined, all except the last one have to be called using 'IA0' as option to TQCE/TQCEL. Once 'IA' is passed for the last incoming amount, the first calculation is made. |
| Blank | - | Calculate without any target variable, VALS is not used. |
| Option | Variable | Default value |
|---|---|---|
| PLOW | Low pressure limit | 10-50 bar |
| PHIGH | High pressure limit | 107 bar |
| VLOW | Low volume limit | 10-7 dm3 |
| VHIGH | High volume limit | 1050 dm3 |
| TLOW | Low temperature limit | 298.15 K |
| THIGH | High temperature limit | 6000 K |
| INDEXP | INDEX | Type of VAL | Components, phases, or constituents for which value is returned |
|---|---|---|---|
| >0 | >0 | DOUBLE PRECISION | Constituent INDEX of phase INDEXP (INDEX=1 for stoichiometric condensed phases), or system component INDEX of phase INDEXP |
| >0 | <0 | Array of DOUBLE PRECISION | All constituents of phase INDEXP, or all system components of phase INDEXP (for options XP and AP) |
| >0 | 0 | DOUBLE PRECISION | Phase INDEXP |
| <0 | 0 | Array of DOUBLE PRECISION | All phases |
| <= 0 | >0 | DOUBLE PRECISION | System component INDEX |
| <= 0 | <0 | Array of DOUBLE PRECISION | All system components |
| 0 | 0 | DOUBLE PRECISION | Entire system |
| Option | Variable | Comment |
|---|---|---|
| P | Total pressure | - |
| VT | Total volume | - |
| T | Temperature | - |
| A | Equilibrium amount | - |
| IA | Incoming amount | Not valid for phases or for the entire system |
MU
AC
|
Chemical potential
Activity/fugacity
|
Relative values for phases and constituents; absolute values for system components; fugacities in current pressure unit for gas phase constituents. |
pH
Eh
|
pH
Eh/V
|
The calculation of pH and Eh/V requires that the constituents H+ in the aqueous phase and H2 in the gas phase be present in the thermochemical data-file. If pH and Eh/V cannot be calculated, the output VAL=0.0 will be returned. In order for ChemApp to recognize H+ and H2 in the data-file, the names of the system components hydrogen and aqueous electron must be 'H' and 'EA', respectively (see TQCNSC). |
CP
H
S
G
V
|
Heat capacity
Enthalpy
Entropy
Gibbs energy
Volume
|
Not valid for system components. Extensive properties for the equilibrium state dependent on the inherent reference state of the data. However, when the entire system is selected and streams are used, the values represent balances, i.e. the extensive property for the equilibrium state minus the sum of values for all streams. To obtain the total volume in this case, use option 'VT'. |
CPM
HM
SM
GM
VM
|
Heat capacity/amount unit
Enthalpy/amount unit
Entropy/amount unit
Gibbs energy/amount unit
Volume/amount unit
|
Not valid for system components. Partial values are returned for constituents, integral values for phases. Values are valid for default or selected amount units. |
| X | Fraction | Mass or mole fraction of a system component in the system, depending on the default or selected amount unit |
| XP | Fraction | Mass or mole fraction of a system component in a phase, depending on the default or selected amount unit |
| AP | Equilibrium amount | Equilibrium amount of system component in a phase |
| Option | Variable |
|---|---|
| CP | Heat capacity |
| H | Enthalpy |
| S | Entropy |
| G | Gibbs energy |
| V | Volume |
| T | Temperature (used to retrieve the upper limit of the current temperature interval for a given phase constituent or a stoichiometric condensed phase) |
| Option | Variable | Unit |
|---|---|---|
| CP | Heat capacity | [current energy unit]/K |
| H | Enthalpy | [current energy unit] |
| S | Entropy | [current energy unit]/K |
| G | Gibbs energy | [current energy unit] |
| V | Volume | [current volume unit] |
| Option | Variable | Comment |
|---|---|---|
PF
PN
|
Total pressure | Pressure is the search variable. The upper and lower bounds of the pressure search interval are in VALS(1) and VALS(2). Use option 'PF' for the first call to TQMAP, 'PN' for all subsequent ones. |
TF
TN
|
Temperature | Temperature is the search variable. The upper and lower bounds of the temperature search interval are in VALS(1) and VALS(2). Use option 'TF' for the first call to TQMAP, 'TN' for all subsequent ones. |
IA0
IAF
IAN
|
Incoming amount | Incoming amount is the search variable. The upper and lower bounds of the composition search interval are in VALS(1) and VALS(2). If incoming amounts for more than one substance need to be defined, all except the last one have to be called using 'IA0' as option to TQMAP. Once 'IAF' is passed for the last incoming amount, the first calculation is made. Use option 'IAN' for all subsequent ones. |
5.1 TQCE
CALCULATE-EQUILIBRIUM
Use TQCE to calculate the equilibrium with current settings of global conditions or streams.
Synopsis
- FORTRAN:
- CALL TQCE(OPTION,INDEXP,INDEXC,VALS,NOERR)
- C:
- tqce(option,indexp,indexc,vals,&noerr);
- Pascal:
- tqce(option,indexp,indexc,vals,noerr);
- Basic:
- Call tqce(option,indexp,indexc,vals,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table State variables for defining targets when calling TQCE/TQCEL |
| INDEXP | INTEGER | Set to the index number for a phase, if necessary |
| INDEXC | INTEGER | Set to the index number for a constituent, if necessary |
| VALS | Array of 2 of DOUBLE PRECISION | Set to an initial estimate of the target variable, or the lower and upper limits of an incoming amount, if necessary |
| NOERR | INTEGER | Returns an error number |
OPTION, INDEXP, INDEX, and VALS are all input. For a calculation without any target, it is sufficient to enter OPTION as a blank character and the remainder as dummies. See Table State variables for defining targets when calling TQCE/TQCEL for the mnemonics of possible target variables, and for the meaning of VALS.
The constraints when two values of VALS entered are: VALS(1) and VALS(2) must not be equal, and VALS(2) must be greater or equal to zero. VALS is entered with the default unit, or a unit selected using TQCSU (see Initialising the Interface and Reading a Data-file). The estimate of the target variable (see Table State variables for defining targets when calling TQCE/TQCEL) need not be particularly accurate. However, unnecessarily long computing times may result if it is far from the equilibrium value. The limits of the target variable can be changed using TQCLIM. While the target variable is specified using TQCE, the target itself must have been set previously using TQSETC or TQSTEC.
! Calculation of different phase equilibria ! Note that this program contains phase target calculations, ! which cannot be performed with the 'light' version of ChemApp PROGRAM CAF18 IMPLICIT NONE INTEGER NOERR, ISLITE, IO2, IC, IN2, ITEMP DOUBLE PRECISION VALS(2), TP(2), RESULT, TEMP ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading. In this case, a data-file containing the ! thermochemical data for the system carbon-nitrogen-oxygen is selected. CALL TQOPNA('cno.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Since we will perform an extensive property target calculation later, ! we will do the equilibrium calculations using streams. This way we can ! associate an initial state regarding temperature and pressure to the ! incoming substances, which will be taken into account when ChemApp ! calculates the extensive property (e.g. heat) balance. ! Since we are going to prefer degrees Celsius as temperature unit, ! we will change the unit now CALL TQCSU('Temperature ', 'C ', NOERR) ! Define a stream at room temperature and 1 bar TP(1) = 20.D0 TP(2) = 1.D0 CALL TQSTTP('inputs ', TP, NOERR) ! Set stream constituents: 0.5 mol of oxygen and 10 grams of carbon ! Get the index number of the phase constituent oxygen ('O2') in the gas ! phase. If a thermochemical data-file for ChemApp contains a gas ! phase, it is always the first one: CALL TQINPC('O2 ', 1, IO2, NOERR) ! Get the index number of the phase carbon ('C') CALL TQINP('C ', IC, NOERR) ! Set the incoming amount of oxygen in the stream (0.5 mol) CALL TQSTCA('inputs ', 1, IO2, .5D0, NOERR) ! Change the amount unit to gram and set the incoming amount of carbon ! in the stream (10 gram) CALL TQCSU('Amount ', 'gram ', NOERR) CALL TQSTCA('inputs ', IC, 0, 10.D0, NOERR) ! Set the temperature for the equilibrium calculation (1500 C) CALL TQSTEC('T ', 0, 1500.D0, NOERR) ! Calculate the equilibrium CALL TQCE(' ', 0, 0, VALS, NOERR) ! Alternatively, TQCEL can be used, which does not only calculate the ! equilibrium, but also writes a ChemSage result table to standard output CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 1500 C
P = 1 bar
V = 122.74 dm3
STREAM CONSTITUENTS AMOUNT/gram TEMPERATURE/C PRESSURE/bar STREAM
O2/GAS/ 1.5999E+01 20.00 1.0000E+00 1
C 1.0000E+01 20.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
CO 6.6514E-01 7.9890E-01 7.9890E-01
CO2 1.6743E-01 2.0110E-01 2.0110E-01
O 3.0441E-09 3.6562E-09 3.6562E-09
O2 1.2593E-09 1.5126E-09 1.5126E-09
C2O 1.6626E-14 1.9970E-14 1.9970E-14
C 2.8498E-17 3.4229E-17 3.4229E-17
O3 9.3779E-22 1.1264E-21 1.1264E-21
C2 2.6273E-22 3.1556E-22 3.1556E-22
C3 9.1060E-25 1.0937E-24 1.0937E-24
C4 2.7548E-33 3.3088E-33 3.3088E-33
C5 2.5090E-36 3.0135E-36 3.0135E-36
TOTAL: 8.3257E-01 1.0000E+00 1.0000E+00
gram ACTIVITY
C 0.0000E+00 2.7932E-04
C_DIAMOND_A4 0.0000E+00 1.4585E-04
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
1.22149E+01 -9.39612E+04 1.16311E+02 -4.58469E+05 1.10558E+02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
3.38382E+01 -9.40697E+04 2.23251E+02 -4.89928E+05 1.22745E+02
Mass fraction of system components:
GAS
C 0.38462
O 0.61538
50 FORMAT(1X,A,G12.5) ! Get the change in volume that takes place CALL TQGETR('V ', 0, 0, RESULT, NOERR) WRITE(*,FMT=50) 'Change in volume/dm^3 is ', RESULT
Change in volume/dm^3 is 110.56
! Get the enthalpy change for the process (since its sign is negative, ! energy is released CALL TQGETR('H ', 0, 0, RESULT, NOERR) WRITE(*,FMT=50) 'Change in enthalpy/J is ', RESULT
Change in enthalpy/J is -93961.
! Get the activity of carbon, which, at 1500 C is far from being unity CALL TQGETR('AC ', IC, 0, RESULT, NOERR) WRITE(*,FMT=50) 'Activity of carbon is ', RESULT
Activity of carbon is 0.27932E-03
! Once an equilibrium has been calculated using TQCE/TQCEL, subsequent ! calculations can be performed using TQCEN/TQCENL, which speeds up the ! equilibrium calculation by taking results from the previous ! calculation as initial estimates DO ITEMP=1510, 1600, 10 TEMP = ITEMP CALL TQSTEC('T ', 0, TEMP, NOERR) CALL TQCEN(' ', 0, 0, VALS, NOERR) CALL TQGETR('AC ', IC, 0, RESULT, NOERR) WRITE(*,*) 'Activity of carbon at T = ', * TEMP, ' C is ', RESULT ENDDO
Activity of carbon at T = 1510.0000000000000 C is 2.6264470995273015E-004 Activity of carbon at T = 1520.0000000000000 C is 2.4714149363797423E-004 Activity of carbon at T = 1530.0000000000000 C is 2.3272087470949939E-004 Activity of carbon at T = 1540.0000000000000 C is 2.1929686051455182E-004 Activity of carbon at T = 1550.0000000000000 C is 2.0679105085828287E-004 Activity of carbon at T = 1560.0000000000000 C is 1.9513190446012930E-004 Activity of carbon at T = 1570.0000000000000 C is 1.8425408207791832E-004 Activity of carbon at T = 1580.0000000000000 C is 1.7409785770743723E-004 Activity of carbon at T = 1590.0000000000000 C is 1.6460861269141717E-004 Activity of carbon at T = 1600.0000000000000 C is 1.5573627473105856E-004
! Similar to TQCEL, TQCENL also provides a ChemSage output table CALL TQCENL(' ', 0, 0, VALS, NOERR)
T = 1600 C
P = 1 bar
V = 129.67 dm3
STREAM CONSTITUENTS AMOUNT/gram TEMPERATURE/C PRESSURE/bar STREAM
O2/GAS/ 1.5999E+01 20.00 1.0000E+00 1
C 1.0000E+01 20.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
CO 6.6514E-01 7.9890E-01 7.9890E-01
CO2 1.6743E-01 2.0110E-01 2.0110E-01
O 2.1027E-08 2.5256E-08 2.5256E-08
O2 9.4832E-09 1.1390E-08 1.1390E-08
C2O 4.0356E-14 4.8472E-14 4.8472E-14
C 2.1347E-16 2.5639E-16 2.5639E-16
O3 3.2996E-20 3.9632E-20 3.9632E-20
C2 1.6278E-21 1.9551E-21 1.9551E-21
C3 3.1116E-24 3.7374E-24 3.7374E-24
C4 1.0919E-32 1.3115E-32 1.3115E-32
C5 5.9958E-36 7.2015E-36 7.2015E-36
TOTAL: 8.3257E-01 1.0000E+00 1.0000E+00
gram ACTIVITY
C 0.0000E+00 1.5574E-04
C_DIAMOND_A4 0.0000E+00 8.1689E-05
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
1.24040E+01 -9.05677E+04 1.18173E+02 -4.80888E+05 1.17480E+02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
3.40272E+01 -9.06762E+04 2.25113E+02 -5.12346E+05 1.29667E+02
Mass fraction of system components:
GAS
C 0.38462
O 0.61538
! Check if we are working with the 'light' version. ! If we do, omit the following phase target calculation(s). CALL TQLITE(ISLITE, NOERR) IF (ISLITE .EQ. 1) THEN WRITE(*,FMT='(3(1X,A,/))') * '*** Phase target calculations have been omitted here,', * '*** since they are not possible with the', * '*** ''light'' version of ChemApp.' ELSE ! PERFORMING target calculations C ! Note that target calculations are not possible with ChemApp "light", ! you need the regular version of ChemApp to run the examples below. C ! Performing target calculations requires that ChemApp needs to be told ! 2 things: ! - the target itself, ! - the target variable, i.e. which variable ChemApp is allowed to vary ! to achieve the target. ! ! First we will perform a formation phase target calculation, which is ! used to determine the conditions under which a phase becomes stable, ! using carbon as an example. As a target variable we will use the ! temperature. The result will be the temperature under which solid ! carbon will just become stable (smoke point). ! Define the target: phase C, amount of C = 0 CALL TQSTEC('A ', IC, 0.D0, NOERR) ! Define the target variable, which is done during the call to TQCE. ! The value passed as VALS(1) will be taken as an initial estimate. VALS(1) = 500.D0 CALL TQCE('T ', 0, 0, VALS, NOERR) ! Get the temperature, which ChemApp determined CALL TQGETR('T ', 0, 0, RESULT, NOERR) WRITE(*,FMT=50) 'Calculated formation temperature of ' // * 'carbon in degrees Celsius: ', RESULT
Calculated formation temperature of carbon in degrees Celsius: 756.35
! Check whether ChemApp calculated correctly by verifying that the ! activity of carbon is unity. CALL TQGETR('AC ', IC, 0, RESULT, NOERR) WRITE(*,FMT=50) 'Activity of carbon is ', RESULT
Activity of carbon is 1.0000
! The equilibrium amount of carbon should still be zero though, as we ! had ChemApp find the temperature where carbon _just_ gets stable CALL TQGETR('A ', IC, 0, RESULT, NOERR) WRITE(*,FMT=50) 'Amount of carbon is ', RESULT
Amount of carbon is 0.0000
! It is also possible to specify a different amount than zero for the ! phase formation target calculation. To to this, pass a different value ! to TQSTEC when defining the target. As an example, calculate the ! temperature where 1 gram of carbon is stable CALL TQSTEC('A ', IC, 1.D0, NOERR) CALL TQCE('T ', 0, 0, VALS, NOERR) CALL TQGETR('T ', 0, 0, RESULT, NOERR) WRITE(*,FMT=50) 'Calculated formation temperature for ' // * '1 gram of carbon in deg. Celsius:', * RESULT
Calculated formation temperature for 1 gram of carbon in deg. Celsius: 713.05
ENDIF
! Before performing extensive property target calculations, TQREMC is
! used to remove all conditions and targets set previously.
CALL TQREMC(-2, NOERR)
! Remove also the stream 'inputs'
CALL TQSTRM('inputs ', NOERR)
! It is also useful to reset VALS, which now still contains a starting
! value for a temperature in VALS(1)
VALS(1) = 0
! If it is also desired to reset all units to default units, which would
! in this case reset the amount to mol and the temperature unit to
! Kelvin, call TQREMC with a value of -1
CALL TQREMC(-1, NOERR)
! Set up a new stream which contains 1 mol of pure oxygen, preheated to
! 400 K at 1 bar
TP(1) = 400.0
TP(2) = 1.0
CALL TQSTTP('oxygen ', TP, NOERR)
CALL TQSTCA('oxygen ', 1, IO2, 1.D0, NOERR)
! Another stream that contains 1 mol of solid carbon at room
! temperature and 1 bar
TP(1) = 298.15
TP(2) = 1.0
CALL TQSTTP('carbon ', TP, NOERR)
CALL TQSTCA('carbon ', IC, 0, 1.D0, NOERR)
! Calculate the equilibrium and retrieve the enthalpy released for this
! process
CALL TQCE(' ', 0, 0, VALS, NOERR)
CALL TQGETR('H ', 0, 0, RESULT, NOERR)
WRITE(*,FMT=50) 'Change in enthalpy/J is ', RESULT
Change in enthalpy/J is -0.36313E+06
! Determine the adiabatic temperature for this process. This would be ! the temperature at which the above mentioned enthalpy is zero ! Define the target: enthalpy, value = 0 CALL TQSTEC('H ', 0, 0.D0, NOERR) ! Define the target variable, which is done during the call to TQCE. ! The value passed as VALS(1) will be taken as an initial estimate. VALS(1) = 1000.D0 CALL TQCE('T ', 0, 0, VALS, NOERR) ! Get the temperature ChemApp calculated (which is the adiabatic ! temperature in this example) CALL TQGETR('T ', 0, 0, RESULT, NOERR) WRITE(*,FMT=50) 'Calculated adiabatic temperature/K for ' // * 'this process: ', RESULT
Calculated adiabatic temperature/K for this process: 3290.4
! One can investigate how this temperature changes if one assumes a ! mixture that resembles air, instead of pure O2. Air has an N2/O2 ratio ! of about 4:1. Keeping 1 mol of oxygen, we thus have to add 4 mol of ! N2. CALL TQINPC('N2 ', 1, IN2, NOERR) CALL TQSTCA('oxygen ', 1, IN2, 4.D0, NOERR) CALL TQCE('T ', 0, 0, VALS, NOERR) CALL TQGETR('T ', 0, 0, RESULT, NOERR) WRITE(*,FMT=50) 'Adiabatic temperature/K for this process ' // * 'with an N2/O2 mixture: ', RESULT END
Adiabatic temperature/K for this process with an N2/O2 mixture: 2295.3
/* Program cac18 */ /* Calculation of different phase equilibria */ /* Note that this program contains phase target calculations, which cannot be performed with the 'light' version of ChemApp */ #include "cacint.h" int main() { LI noerr, islite, iO2, iC, iN2; DB vals[2], TP[2], result, temp; /* Initialise ChemApp */ tqini(&noerr); /* Open data-file for reading. In this case, a data-file containing the thermochemical data for the system carbon-nitrogen-oxygen is selected. */ tqopna("cno.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Since we will perform an extensive property target calculation later, we will do the equilibrium calculations using streams. This way we can associate an initial state regarding temperature and pressure to the incoming substances, which will be taken into account when ChemApp calculates the extensive property (e.g. heat) balance. */ /* Since we are going to prefer degrees Celsius as temperature unit, we will change the unit now */ tqcsu("Temperature", "C", &noerr); /* Define a stream at room temperature and 1 bar */ TP[0] = 20.0; TP[1] = 1.0; tqsttp("inputs", TP, &noerr); /* Set stream constituents: 0.5 mol of oxygen and 10 grams of carbon */ /* Get the index number of the phase constituent oxygen ("O2") in the gas phase. If a thermochemical data-file for ChemApp contains a gas phase, it is always the first one: */ tqinpc("O2", 1, &iO2, &noerr); /* Get the index number of the phase carbon ("C") */ tqinp("C", &iC, &noerr); /* Set the incoming amount of oxygen in the stream (0.5 mol) */ tqstca("inputs", 1, iO2, .5, &noerr); /* Change the amount unit to gram and set the incoming amount of carbon in the stream (10 gram) */ tqcsu("Amount", "gram", &noerr); tqstca("inputs", iC, 0, 10.0, &noerr); /* Set the temperature for the equilibrium calculation (1500 C) */ tqstec("T", 0, 1500.0, &noerr); /* Calculate the equilibrium */ tqce(" ", 0, 0, vals, &noerr); /* Get the change in volume that takes place */ tqgetr("V", 0, 0, &result, &noerr); printf("Change in volume/dm^3 is %g\n", result);
Change in volume/dm^3 is 110.558
/* Get the enthalpy change for the process (since its sign is negative, energy is released */ tqgetr("H", 0, 0, &result, &noerr); printf("Change in enthalpy/J is %g\n", result);
Change in enthalpy/J is -93961.2
/* Get the activity of carbon, which, at 1500 C is far from being unity */ tqgetr("ac", iC, 0, &result, &noerr); printf("Activity of carbon is %g\n", result);
Activity of carbon is 0.000279325
/* Once an equilibrium has been calculated using tqce/tqcel, subsequent calculations can be performed using tqcen/tqcenl, which speeds up the equilibrium calculation by taking results from the previous calculation as initial estimates */ for (temp = 1510; temp <= 1600; temp +=10) { tqstec("T", 0, temp, &noerr); tqcen(" ", 0, 0, vals, &noerr); tqgetr("ac", iC, 0, &result, &noerr); printf("Activity of carbon at T = %g C is %g\n", temp, result); }
Activity of carbon at T = 1510 C is 0.000262645 Activity of carbon at T = 1520 C is 0.000247141 Activity of carbon at T = 1530 C is 0.000232721 Activity of carbon at T = 1540 C is 0.000219297 Activity of carbon at T = 1550 C is 0.000206791 Activity of carbon at T = 1560 C is 0.000195132 Activity of carbon at T = 1570 C is 0.000184254 Activity of carbon at T = 1580 C is 0.000174098 Activity of carbon at T = 1590 C is 0.000164609 Activity of carbon at T = 1600 C is 0.000155736
/* Similar to tqcel, tqcenl also provides a ChemSage output table */ tqcenl(" ", 0, 0, vals, &noerr);
T = 1600 C
P = 1 bar
V = 129.67 dm3
STREAM CONSTITUENTS AMOUNT/gram TEMPERATURE/C PRESSURE/bar STREAM
O2/GAS/ 1.5999E+01 20.00 1.0000E+00 1
C 1.0000E+01 20.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
CO 6.6514E-01 7.9890E-01 7.9890E-01
CO2 1.6743E-01 2.0110E-01 2.0110E-01
O 2.1027E-08 2.5256E-08 2.5256E-08
O2 9.4832E-09 1.1390E-08 1.1390E-08
C2O 4.0356E-14 4.8472E-14 4.8472E-14
C 2.1347E-16 2.5639E-16 2.5639E-16
O3 3.2996E-20 3.9632E-20 3.9632E-20
C2 1.6278E-21 1.9551E-21 1.9551E-21
C3 3.1116E-24 3.7374E-24 3.7374E-24
C4 1.0919E-32 1.3115E-32 1.3115E-32
C5 5.9958E-36 7.2015E-36 7.2015E-36
TOTAL: 8.3257E-01 1.0000E+00 1.0000E+00
gram ACTIVITY
C 0.0000E+00 1.5574E-04
C_DIAMOND_A4 0.0000E+00 8.1689E-05
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
1.24040E+01 -9.05677E+04 1.18173E+02 -4.80888E+05 1.17480E+02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
3.40272E+01 -9.06762E+04 2.25113E+02 -5.12346E+05 1.29667E+02
Mass fraction of system components:
GAS
C 0.38462
O 0.61538
/* Check if we are working with the 'light' version. If we do, omit the following phase target calculation(s). */ tqlite(&islite, &noerr); if (islite) { printf("*** Phase target calculations have been omitted here,\n" "*** since they are not possible with the\n" "*** 'light' version of ChemApp.\n\n"); } else { /* Performing target calculations */ /* Note that target calculations are not possible with ChemApp "light", you need the regular version of ChemApp to run the examples below. */ /* Performing target calculations requires that ChemApp needs to be told 2 things: - the target itself, - the target variable, i.e. which variable ChemApp is allowed to vary to achieve the target. First we will perform a formation phase target calculation, which is used to determine the conditions under which a phase becomes stable, using carbon as an example. As a target variable we will use the temperature. The result will be the temperature under which solid carbon will just become stable (smoke point). */ /* Define the target: phase C, amount of C = 0 */ tqstec("a", iC, 0.0, &noerr); /* Define the target variable, which is done during the call to tqce. The value passed as vals[0] will be taken as an initial estimate. */ vals[0] = 500.0; tqce("T", 0, 0, vals, &noerr); /* Get the temperature, which ChemApp determined */ tqgetr("T", 0, 0, &result, &noerr); printf("Calculated formation temperature of " "carbon in degrees Celsius: %g\n", result);
Calculated formation temperature of carbon in degrees Celsius: 756.345
/* Check whether ChemApp calculated correctly by verifying that the activity of carbon is unity. */ tqgetr("ac", iC, 0, &result, &noerr); printf("Activity of carbon is %g\n", result);
Activity of carbon is 1
/* The equilibrium amount of carbon should still be zero though, as we had ChemApp find the temperature where carbon _just_ gets stable */ tqgetr("a", iC, 0, &result, &noerr); printf("Amount of carbon is %g\n", result);
Amount of carbon is 0
/* It is also possible to specify a different amount than zero for the phase formation target calculation. To to this, pass a different value to tqstec when defining the target. As an example, calculate the temperature where 1 gram of carbon is stable */ tqstec("a", iC, 1.0, &noerr); tqce("T", 0, 0, vals, &noerr); tqgetr("T", 0, 0, &result, &noerr); printf("Calculated formation temperature for " "1 gram of carbon in deg. Celsius: %g\n", result);
Calculated formation temperature for 1 gram of carbon in deg. Celsius: 713.054
} /* Before performing extensive property target calculations, tqremc is used to remove all conditions and targets set previously */ tqremc(-2, &noerr); /* Remove also the stream "inputs" */ tqstrm("inputs", &noerr); /* It is also useful to reset vals, which now still contains a starting value for a temperature in vals[0] */ vals[0] = 0; /* If it is also desired to reset all units to default units, which would in this case reset the amount to mol and the temperature unit to Kelvin, call tqremc with a value of -1 */ tqremc(-1, &noerr); /* Set up a new stream which contains 1 mol of pure oxygen, preheated to 400 K at 1 bar */ TP[0] = 400.0; TP[1] = 1.0; tqsttp("oxygen", TP, &noerr); tqstca("oxygen", 1, iO2, 1.0, &noerr); /* Another stream that contains 1 mol of solid carbon at room temperature and 1 bar */ TP[0] = 298.15; TP[1] = 1.0; tqsttp("carbon", TP, &noerr); tqstca("carbon", iC, 0, 1.0, &noerr); /* Calculate the equilibrium and retrieve the enthalpy released for this process */ tqce(" ", 0, 0, vals, &noerr); tqgetr("H", 0, 0, &result, &noerr); printf("Change in enthalpy/J is %g\n", result);
Change in enthalpy/J is -363127
/* Determine the adiabatic temperature for this process. This would be the temperature at which the above mentioned enthalpy is zero */ /* Define the target: enthalpy, value = 0 */ tqstec("H", 0, 0.0, &noerr); /* Define the target variable, which is done during the call to tqce. The value passed as vals[0] will be taken as an initial estimate. */ vals[0] = 1000.0; tqce("T", 0, 0, vals, &noerr); /* Get the temperature ChemApp calculated (which is the adiabatic temperature in this example) */ tqgetr("T", 0, 0, &result, &noerr); printf("Calculated adiabatic temperature/K for " "this process: %g\n", result);
Calculated adiabatic temperature/K for this process: 3290.38
/* One can investigate how this temperature changes if one assumes a mixture that resembles air, instead of pure O2. Air has an N2/O2 ratio of about 4:1. Keeping 1 mol of oxygen, we thus have to add 4 mol of N2. */ tqinpc("N2", 1, &iN2, &noerr); tqstca("oxygen", 1, iN2, 4.0, &noerr); tqce("T", 0, 0, vals, &noerr); tqgetr("T", 0, 0, &result, &noerr); printf("Adiabatic temperature/K for this process " "with a N2/O2 mixture: %g\n", result); return 0; }
Adiabatic temperature/K for this process with a N2/O2 mixture: 2295.3
5.2 TQCEL
CALCULATE-EQUILIBRIUM-AND-LIST-RESULTS
Use TQCEL to calculate the equilibrium with current settings of global conditions or streams, and list the results in the form of a ChemSage format output table.
Synopsis
- FORTRAN:
- CALL TQCEL(OPTION,INDEXP,INDEXC,VALS,NOERR)
- C:
- tqcel(option,indexp,indexc,vals,&noerr);
- Pascal:
- tqcel(option,indexp,indexc,vals,noerr);
- Basic:
- Call tqcel(option,indexp,indexc,vals,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table State variables for defining targets when calling TQCE/TQCEL |
| INDEXP | INTEGER | Set to the index number for a phase, if necessary |
| INDEXC | INTEGER | Set to the index number for a constituent, if necessary |
| VALS | Array of 2 of DOUBLE PRECISION | Set to an initial estimate of the target variable, or the lower and upper limits of an incoming amount, if necessary |
| NOERR | INTEGER | Returns an error number |
The parameters and their conditions are identical to those for TQCE. A result table is produced after finishing the calculations and is written to unit LIST, see Table Legal input/output options used by TQGIO and TQCIO. The amount unit used in the output table is the one which is currently selected (i.e. 'mol' by default).
- See
- TQCE
5.3 TQCEN
CALCULATE-EQUILIBRIUM-NO-ESTIMATION
Use TQCEN to calculate the equilibrium, taking results from the previous equilibrium calculation as initial estimates
Added for ChemApp version 5.0.0
Synopsis
- FORTRAN:
- CALL TQCEN(OPTION,INDEXP,INDEXC,VALS,NOERR)
- C:
- tqcen(option,indexp,indexc,vals,&noerr);
- Pascal:
- tqcen(option,indexp,indexc,vals,noerr);
- Basic:
- Call tqcen(option,indexp,indexc,vals,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table State variables for defining targets when calling TQCE/TQCEL |
| INDEXP | INTEGER | Set to the index number for a phase, if necessary |
| INDEXC | INTEGER | Set to the index number for a constituent, if necessary |
| VALS | Array of 2 of DOUBLE PRECISION | Set to an initial estimate of the target variable, or the lower and upper limits of an incoming amount, if necessary |
| NOERR | INTEGER | Returns an error number |
Once an equilibrium has been calculated with TQCE or TQCEL, subsequent equilibria can be executed using TQCEN or TQCENL. When the two latter subroutines are called, results from the previous equilibrium calculation are used as initial estimates. This will result in a noticable increase of the computational efficiency, especially when the current settings of global conditions or streams are close to those of the previous calculation.
Error code 516 will result if the status of a phase constituent or a phase has been modified since the last executed phase equilibrium. Thus, after a call to TQCEN, the application programmer is advised to check NOERR for this value, and recalculate the equilibrium using TQCE if it equals 516.
For systems which include mixtures described with the models SUBG, SUBS, or QUAS (see Table Identifiers of phase model names as returned by TQMODL), the use of TQCEN might not necessarily lead to shorter computing times.
If convergence would fail because of poor initial estimates, or if a system component is added or deleted after the last executed equilibrium, then the calculation will be automatically repeated with initial estimates produced by ChemApp.
For a description of the parameters and constraints, see TQCE.
- See
- TQCE
5.4 TQCENL
CALCULATE-EQUILIBRIUM-NO-ESTIMATES-AND-LIST-RESULTS
Use TQCENL to calculate the equilibrium, taking results from the previous equilibrium calculation as initial estimates, and list the results in the form of a ChemSage format output table.
Added for ChemApp version 5.0.0
Synopsis
- FORTRAN:
- CALL TQCENL(OPTION,INDEXP,INDEXC,VALS,NOERR)
- C:
- tqcenl(option,indexp,indexc,vals,&noerr);
- Pascal:
- tqcenl(option,indexp,indexc,vals,noerr);
- Basic:
- Call tqcenl(option,indexp,indexc,vals,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table State variables for defining targets when calling TQCE/TQCEL |
| INDEXP | INTEGER | Set to the index number for a phase, if necessary |
| INDEXC | INTEGER | Set to the index number for a constituent, if necessary |
| VALS | Array of 2 of DOUBLE PRECISION | Set to an initial estimate of the target variable, or the lower and upper limits of an incoming amount, if necessary |
| NOERR | INTEGER | Returns an error number |
The parameters and their conditions are identical to those for TQCE, TQCEL, and TQCEN. A result table is produced after finishing the calculations and is written to unit LIST, see Table Legal input/output options used by TQGIO and TQCIO. The amount unit used in the output table is the one which is currently selected (i.e. 'mol' by default).
See the description for TQCEN with respect to possible errors messages returned by TQCENL.
- See
- TQCE
5.5 TQMAP
CALCULATE-ONE-DIMENSIONAL-PHASE-MAP
Use TQMAP to perform a one-dimensional phase mapping calculation, giving all phase transitions within a defined interval.
Added for ChemApp version 3.0.0
Synopsis
- FORTRAN:
- CALL TQMAP(OPTION,INDEXP,INDEXC,VALS,ICONT,NOERR)
- C:
- tqmap(option,indexp,indexc,vals,&icont,&noerr);
- Pascal:
- tqmap(option,indexp,indexc,vals,icont,noerr);
- Basic:
- Call tqmap(option,indexp,indexc,vals,icont,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table State variables for defining one-dimensional phase mapping calculations using TQMAP/TQMAPL |
| INDEXP | INTEGER | Set to the index number for a phase, if necessary |
| INDEXC | INTEGER | Set to the index number for a constituent, if necessary |
| VALS | Array of 2 of DOUBLE PRECISION | Set to the lower and upper limits of the search variable |
| ICONT | INTEGER | Returns a value indicating whether more calls to TQMAP/TQMAPL are necessary |
| NOERR | INTEGER | Returns an error number |
OPTION, INDEXP, INDEXC, and VALS are all input, ICONT and NOERR are output. See Table State variables for defining one-dimensional phase mapping calculations using TQMAP/TQMAPL for the mnemonics of possible search variables. VALS is an array of two double precision values which is always used to pass the lower and upper bounds of the search interval to TQMAP.
The constraints for the two values of VALS entered are: VALS(1) and VALS(2) must not be equal, and VALS(2) must be greater than, or, for incoming amount as search variable, equal to zero. VALS is entered with the default unit, or a unit set using TQCSU (see Initialising the Interface and Reading a Data-file).
For every one-dimensional phase map, the first two calls to TQMAP return the equilibrium state of the system at VALS(1) and VALS(2) and not results relating to any phase boundary. Beginning with the third and every subsequent call of TQMAP, the output variable ICONT has to be tested. If it does have a positive value, more calls to TQMAP need to be made.
In case incoming amount is the search variable, and incoming amounts for more than one substance need to be defined, all except the last one have to be called using 'IA0' as option to TQMAP. Once 'IAF' is passed for the last incoming amount, the first calculation is made. Subsequently 'IAN' is used as OPTION for all following calls to TQMAP. See One-dimensional phase mapping for an example of how to use TQMAP with incoming amount as search variable.
! One-dimensional phase mapping calculation ! Note that this program contains phase target calculations, ! which cannot be performed with the 'light' version of ChemApp PROGRAM CAF24 IMPLICIT NONE INTEGER NOERR, NUMCON, ISLITE, ISIO2, RESNO, ICONT DOUBLE PRECISION VALS(2), RESULT RESNO = 0 ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Check if we are working with the 'light' version. ! If we do, omit the following phase target calculation(s). CALL TQLITE(ISLITE, NOERR) IF (ISLITE .EQ. 1) THEN WRITE(*,FMT='(3(1X,A,/))') * '*** Phase target calculations have been omitted here,', * '*** since they are not possible with the', * '*** ''light'' version of ChemApp.' ELSE ! Determine the index number for the phase SiO2(quartz) CALL TQINP('SiO2(quartz) ', ISIO2, NOERR) ! Enter one mol of SiO2 CALL TQSETC('IA ', ISIO2, 0, 1.0D0, NUMCON, NOERR) ! The temperature search interval is supposed to range from 300 to ! 3000 K: VALS(1) = 300.D0 VALS(2) = 3000.D0 ! First call to TQMAP, note the 'F' ('First') in the option parameter CALL TQMAP('TF ',0,0,VALS,ICONT,NOERR) ! The variable RESNO keeps track of the number of times we called TQMAP: RESNO = RESNO + 1 ! Retrieve and print the temperature, which we know is VALS(1) CALL TQGETR('T ',0,0,RESULT,NOERR) 100 FORMAT(A,F8.2,A) WRITE(*,100) '*** Lower interval boundary:', RESULT, ' K' 30 CONTINUE ! TQMAP is called again. Note the 'N' ('Next') in the option ! parameter. If we are at the first phase boundary (RESNO is still 2), ! we call TQMAPL for a change to produce a ChemSage output table... IF (RESNO .EQ. 2) THEN WRITE(*,FMT='(A)') * '*** ChemSage result table for the first phase boundary found:' CALL TQMAPL('TN ',0,0,VALS,ICONT,NOERR) WRITE(*,*) ! ...otherwise we just call TQMAP: ELSE CALL TQMAP('TN ',0,0,VALS,ICONT,NOERR) ENDIF RESNO = RESNO +1 ! Get the temperature... CALL TQGETR('T ',0,0,RESULT,NOERR) ! ... and print the entry for the result table. If we have called TQMAP ! twice already, we know that we have found a phase boundary. If not, we ! have retrieved the temperature value of the upper interval boundary ! VALS(2): IF (RESNO .GT. 2) THEN WRITE(*,100) '*** Phase boundary found at ', RESULT, ' K' ELSE WRITE(*,100) '*** Upper interval boundary: ', RESULT, ' K' ENDIF ! For as long as ICONT is positive, we need to make further calls of ! TQMAP IF (ICONT .GT. 0) GOTO 30
*** Lower interval boundary: 300.00 K
*** Upper interval boundary: 3000.00 K
*** ChemSage result table for the first phase boundary found:
*T = 1140.10 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
SiO2(quartz) 1.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
SiO 0.0000E+00 6.2502E-01 4.4829E-16
O2 0.0000E+00 2.5088E-01 1.7994E-16
O 0.0000E+00 1.2327E-01 8.8415E-17
SiO2 0.0000E+00 8.3208E-04 5.9681E-19
Si 0.0000E+00 4.4651E-15 3.2026E-30
O3 0.0000E+00 2.4636E-19 1.7670E-34
Si2 0.0000E+00 5.5659E-36 3.9921E-51
Si3 0.0000E+00 9.0161E-54 6.4667E-69
TOTAL: 0.0000E+00 1.0000E+00 7.1725E-16
mol ACTIVITY
SiO2(quartz) 1.0000E+00 1.0000E+00
SiO2(tridymite) 0.0000E+00 1.0000E+00
SiO2(cristobali) 0.0000E+00 9.9398E-01
SiO2(liquid) 0.0000E+00 6.4439E-01
Si 0.0000E+00 2.5802E-17
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
7.04990E+01 -8.55398E+05 1.25413E+02 -9.98382E+05 0.00000E+00
*** Phase boundary found at 1140.10 K
*** Phase boundary found at 1738.28 K
*** Phase boundary found at 1995.99 K
! With the above example the temperatures of all phase boundaries in a ! system which contains 1 mol of SiO2 have been calculated. Thus the ! phase boundaries determined reflect the stability ranges of the ! various modifications of SiO2. Also note again that the first two ! temperatures determined are _no_ phase boundaries, but the lower and ! upper limit of the search interval (VALS(1) and VALS(2)). ! ! For the first phase boundary found, TQMAPL is called instead of TQMAP, ! thus producing a ChemSage output table. From this ChemSage output ! table it can be seen that at the temperature calculated SiO2 ! transforms from the quartz into the tridymite modification. ! ! Refer to the worked example 5 (One-dimensional phase mapping) for a ! further example. This also demonstrates how to obtain information on ! which phases are stable at each of the phase boundaries determined. ENDIF END
/* Program cac24 */ /* One-dimensional phase mapping calculation */ /* Note that this program contains phase target calculations, which cannot be performed with the 'light' version of ChemApp */ #include "cacint.h" int main() { LI noerr, numcon, islite, iSiO2, resno=0, icont; DB vals[2], result; /* Initialise ChemApp */ tqini(&noerr); /* Open data-file for reading. */ tqopna("cosi.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Check if we are working with the 'light' version. If we do, omit the following phase target calculation(s). */ tqlite(&islite, &noerr); if (islite) { printf("*** Phase target calculations have been omitted here,\n" "*** since they are not possible with the\n" "*** 'light' version of ChemApp.\n\n"); } else { /* Determine the index number for the phase SiO2(quartz) */ tqinp("SiO2(quartz)", &iSiO2, &noerr); /* Enter one mol of SiO2 */ tqsetc("IA", iSiO2, 0, 1.0, &numcon, &noerr); /* The temperature search interval is supposed to range from 300 to 3000 K: */ vals[0] = 300.0; vals[1] = 3000.0; /* First call to tqmap, note the "f" ("first") in the option parameter */ tqmap("tf", 0, 0, vals, &icont, &noerr); /* The variable resno keeps track of the number of times we call tqmap: */ resno++; /* Retrieve and print the temperature, which we know is vals[0]: */ tqgetr("t", 0, 0, &result, &noerr); printf("*** Lower interval boundary: %g K\n", result); /* For as long as icont is positive, we need to make further calls of tqmap */ while (icont) { /* tqmap is called again. Note the "n" ("next") in the option parameter. If we are at the first phase boundary (resno is still 2), we call tqmapl for a change to produce a ChemSage output table... */ if (resno == 2) { printf("*** ChemSage result table " "for the first phase boundary found:\n"); tqmapl("tn", 0, 0, vals, &icont, &noerr); printf("\n"); /* ...otherwise we just call tqmap: */ } else { tqmap("tn", 0, 0, vals, &icont, &noerr); } resno++; /* Get the temperature... */ tqgetr("t", 0, 0, &result, &noerr); /* ...and print the entry for the result table. If we have called tqmap twice already, we know that we have found a phase boundary. If not, we have retrieved the temperature value of the upper interval boundary (vals[1]): */ if (resno > 2) { printf("*** Phase boundary found at %g K\n", result); } else { printf("*** Upper interval boundary: %g K\n", result); } }
*** Lower interval boundary: 300 K
*** Upper interval boundary: 3000 K
*** ChemSage result table for the first phase boundary found:
*T = 1140.10 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
SiO2(quartz) 1.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
SiO 0.0000E+00 6.2502E-01 4.4829E-16
O2 0.0000E+00 2.5088E-01 1.7994E-16
O 0.0000E+00 1.2327E-01 8.8415E-17
SiO2 0.0000E+00 8.3208E-04 5.9681E-19
Si 0.0000E+00 4.4651E-15 3.2026E-30
O3 0.0000E+00 2.4636E-19 1.7670E-34
Si2 0.0000E+00 5.5659E-36 3.9921E-51
Si3 0.0000E+00 9.0161E-54 6.4667E-69
TOTAL: 0.0000E+00 1.0000E+00 7.1725E-16
mol ACTIVITY
SiO2(quartz) 1.0000E+00 1.0000E+00
SiO2(tridymite) 0.0000E+00 1.0000E+00
SiO2(cristobali) 0.0000E+00 9.9398E-01
SiO2(liquid) 0.0000E+00 6.4439E-01
Si 0.0000E+00 2.5802E-17
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
7.04990E+01 -8.55398E+05 1.25413E+02 -9.98382E+05 0.00000E+00
*** Phase boundary found at 1140.1 K
*** Phase boundary found at 1738.28 K
*** Phase boundary found at 1995.99 K
/* With the above example the temperatures of all phase boundaries in a system which contains 1 mol of SiO2 have been calculated. Thus the phase boundaries determined reflect the stability ranges of the various modifications of SiO2. Also note that the first two temperatures determined are _no_ phase boundaries, but the lower and upper limit of the search interval (vals[0] and vals[1]). For the first phase boundary found, tqmapl is called instead of tqmap, thus producing a ChemSage output table. From this ChemSage output table it can be seen that at the temperature calculated SiO2 transforms from the quartz into the tridymite modification. Refer to the worked example 5 (One-dimensional phase mapping) for a further example. This also demonstrates how to obtain information on which phases are stable at each of the phase boundaries determined. */ } return 0; }
5.6 TQMAPL
CALCULATE-ONE-DIMENSIONAL-PHASE-MAP-AND-LIST-RESULTS
Use TQMAP to perform a one-dimensional phase mapping calculation and list the results in the form of a ChemSage format output table.
Added for ChemApp version 3.0.0
Synopsis
- FORTRAN:
- CALL TQMAPL(OPTION,INDEXP,INDEXC,VALS,ICONT,NOERR)
- C:
- tqmapl(option,indexp,indexc,vals,&icont,&noerr);
- Pascal:
- tqmapl(option,indexp,indexc,vals,icont,noerr);
- Basic:
- Call tqmapl(option,indexp,indexc,vals,icont,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table State variables for defining one-dimensional phase mapping calculations using TQMAP/TQMAPL |
| INDEXP | INTEGER | Set to the index number for a phase, if necessary |
| INDEXC | INTEGER | Set to the index number for a constituent, if necessary |
| VALS | Array of 2 of DOUBLE PRECISION | Set to the lower and upper limits of the search variable |
| ICONT | INTEGER | Returns a value indicating whether more calls to TQMAP / TQMAPL are necessary |
| NOERR | INTEGER | Returns an error number |
The parameters and their conditions are identical to those for TQMAP, except that a result table is produced after conclusion of the calculation. This output is written to unit LIST, see Table Legal input/output options used by TQGIO and TQCIO. The amount unit used in the output table is the one which is currently selected (i.e. 'mol' by default).
- See
- TQMAP
5.7 TQCLIM
CHANGE-LIMIT-OF-TARGET-VARIABLE
Use TQCLIM to modify the upper and lower limits of the target variables pressure, volume, or temperature.
Synopsis
- FORTRAN:
- CALL TQCLIM(OPTION,VAL,NOERR)
- C:
- tqclim(option,val,&noerr);
- Pascal:
- tqclim(option,val,noerr);
- Basic:
- Call tqclim(option,val,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table Options for modifying upper and lower limits of target variables using TQCLIM |
| VAL | DOUBLE PRECISION | Set to a limit in the default unit |
| NOERR | INTEGER | Returns an error number |
OPTION is a character variable identifying the limit to be changed. VAL is entered with a default unit (bar/dm3/K), irrespective of which system units have been set with TQCSU. In a target calculation, the lower and upper limits of a target variable are never exceeded. One specific solution of several possible ones can be found by changing the default numbers in Table Options for modifying upper and lower limits of target variables using TQCLIM, such that the interval is reduced. By reducing TLOW, a calculated temperature can have a value below room temperature. When composition is the target variable, the range can only be changed by modifying VALS(1) and VALS(2) using TQCE/TQCEL, see Table State variables for defining targets when calling TQCE/TQCEL.
! Change the limits of target variables ! Note that this program contains phase target calculations, ! which cannot be performed with the 'light' version of ChemApp PROGRAM CAF22 IMPLICIT NONE INTEGER NOERR, ISLITE, NUMCON, IPB, ISN, ILIQ, IFCC DOUBLE PRECISION TEMP, ACT, VALS(2) ! Initialise ChemApp CALL TQINI(NOERR) ! Check if we are working with the 'light' version. ! If we do, omit the following phase target calculation(s). CALL TQLITE(ISLITE, NOERR) IF (ISLITE .EQ. 1) THEN WRITE(*,FMT='(3(1X,A,/))') * '*** Phase target calculations have been omitted here,', * '*** since they are not possible with the', * '*** ''light'' version of ChemApp.' ELSE ! Open data-file for reading CALL TQOPNA('pbsn.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) 50 FORMAT(1X,A,G12.5,A) 51 FORMAT(1X,2(A,G12.5)) ! For this example, let us assume one intends to find the solidus point ! for an Pb-Sn alloy consisting of 20 mol-% Sn and 80 mol-% Pb by ! performing a precipitation target calculation from the FCC ! phase. Figure ref(pbsn2_pd) shows the Pb-Sn phase diagram as ! calculated by ChemSage. The line marked "1" indicates the ! concentration that will be used for the precipitation target ! calculations below, using the temperature as the target variable. ! Get index numbers of system components CALL TQINSC('Pb ', IPB, NOERR) CALL TQINSC('Sn ', ISN, NOERR) ! Get index number of the FCC and liquid phases CALL TQINP('FCC ', IFCC, NOERR) CALL TQINP('LIQ ', ILIQ, NOERR) ! Input 0.2 mol Sn and 0.8 mol Pb CALL TQSETC('IA ', 0, ISN, 0.2D0, NUMCON, NOERR) CALL TQSETC('IA ', 0, IPB, 0.8D0, NUMCON, NOERR) ! Define the precipitation phase target CALL TQSETC('A ', IFCC, 0, -1.D0, NUMCON, NOERR) ! Define the target variable (temperature) and perform the ! equilibrium calculation. The estimate for the target variable, passed ! via VALS(1), is set to 400 K. VALS(1) = 400.D0 CALL TQCE('T ', 0, 0, VALS, NOERR) ! Retrieve the calculated temperature CALL TQGETR('T ', 0, 0, TEMP, NOERR) WRITE(*,FMT=50) 'Calculated solidus temperature: ', * TEMP, 'K'
Calculated solidus temperature: 426.36 K
! Looking at the Pb-Sn phase diagram, this temperature is certainly ! wrong. This can be demonstrated by retrieving the activity of the ! liquid phase: CALL TQGETR('AC ', ILIQ, 0, ACT, NOERR) WRITE(*,FMT=51) 'Activity of liquid phase at ',TEMP, ' K: ', * ACT
Activity of liquid phase at 426.36 K: 0.89170
! Since the activity is less than unity, the liquid phase is not stable, ! thus this cannot be the solidus point. The reason is that ChemApp ! found the phase boundary between FCC and the two-phase region FCC+BCT, ! instead of that between FCC and FCC+Liquid. ! To keep ChemApp from finding the unwanted phase boundary at lower ! temperatures, tell it to limit its search to temperatures above 450 K: CALL TQCLIM('TLOW ', 450.D0, NOERR) ! Recalculate the equilibrium CALL TQCE('T ', 0, 0, VALS, NOERR) ! Retrieve the calculated temperature CALL TQGETR('T ', 0, 0, TEMP, NOERR) WRITE(*,FMT=50) 'Calculated solidus temperature: ', * TEMP, 'K'
Calculated solidus temperature: 505.37 K
! Check the activity of the liquid phase again to verify that it is ! indeed stable: CALL TQGETR('AC ', ILIQ, 0, ACT, NOERR) WRITE(*,FMT=51) 'Activity of liquid phase at ',TEMP, ' K: ', * ACT ! Since the activity is unity, we know that the temperature we found is ! correct. ENDIF END
Activity of liquid phase at 505.37 K: 1.0000
/* Program cac22 */ /* Change the limits of target variables */ /* Note that this program contains phase target calculations, which cannot be performed with the 'light' version of ChemApp */ #include "cacint.h" int main() { LI noerr, islite, numcon, iPb, iSn, iLIQ, iFCC; DB temp, act, vals[2]; /* Initialise ChemApp */ tqini(&noerr); /* Check if we are working with the 'light' version. If we do, omit the following phase target calculation(s). */ tqlite(&islite, &noerr); if (islite) { printf("*** Phase target calculations have been omitted here,\n" "*** since they are not possible with the\n" "*** 'light' version of ChemApp.\n\n"); } else { /* Open data-file for reading */ tqopna("pbsn.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* For this example, let us assume one intends to find the solidus point for an Pb-Sn alloy consisting of 20 mol-% Sn and 80 mol-% Pb by performing a precipitation target calculation from the FCC phase. Figure ref(pbsn2_pd) shows the Pb-Sn phase diagram as calculated by ChemSage. The line marked "1" indicates the concentration that will be used for the precipitation target calculations below, using the temperature as the target variable. */ /* Get index numbers of system components */ tqinsc("Pb", &iPb, &noerr); tqinsc("Sn", &iSn, &noerr); /* Get phase index numbers of the FCC and liquid phases */ tqinp("FCC", &iFCC, &noerr); tqinp("LIQ", &iLIQ, &noerr); /* Input 0.2 mol Sn and 0.8 mol Pb */ tqsetc("ia", 0, iSn, 0.2, &numcon, &noerr); tqsetc("ia", 0, iPb, 0.8, &numcon, &noerr); /* Define the precipitation target */ tqsetc("a", iFCC, 0, -1.0, &numcon, &noerr); /* Define the target variable (temperature) and perform the equilibrium calculation. The estimate for the target variable, passed C via vals[0], is set to 400 K. */ vals[0] = 400.0; tqce("T", 0, 0, vals, &noerr); /* Retrieve the calculated temperature */ tqgetr("T", 0, 0, &temp, &noerr); printf("Calculated solidus temperature: %g K\n", temp);
Calculated solidus temperature: 426.36 K
/* Looking at the Pb-Sn phase diagram, this temperature is certainly wrong. This can be demonstrated by retrieving the activity of the liquid phase: */ tqgetr("ac", iLIQ, 0, &act, &noerr); printf("Activity of liquid phase at %g K: %g\n", temp, act);
Activity of liquid phase at 426.36 K: 0.891702
/* Since the activity is less than unity, the liquid phase is not stable, thus this cannot be the solidus point. The reason is that ChemApp found the phase boundary between FCC and the two-phase region FCC+BCT, instead of that between FCC and FCC+Liquid. */ /* To keep ChemApp from finding the unwanted phase boundary at lower temperatures, tell it to limit its search to temperatures above 450 K: */ tqclim("TLOW", 450.0, &noerr); /* Recalculate the equilibrium */ tqce("T", 0, 0, vals, &noerr); /* Retrieve the calculated temperature */ tqgetr("T", 0, 0, &temp, &noerr); printf("Calculated solidus temperature: %g K\n", temp);
Calculated solidus temperature: 505.369 K
/* Check the activity of the liquid phase again to verify that it is indeed stable: */ tqgetr("ac", iLIQ, 0, &act, &noerr); printf("Activity of liquid phase at %g K: %g\n", temp, act); /* Since the activity is unity, we know that the temperature we found is correct. */ } return 0; }
Activity of liquid phase at 505.369 K: 1
5.8 TQSHOW
SHOW-PRESENT-SETTINGS
Use TQSHOW to display the present settings.
Synopsis
- FORTRAN:
- CALL TQSHOW(NOERR)
- C:
- tqshow(&noerr);
- Pascal:
- tqshow(noerr);
- Basic:
- Call tqshow(noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| NOERR | INTEGER | Returns an error number |
TQSHOW displays information about the currently set conditions and streams, and is useful for debugging. Its output is stored on the unit/file associated with LIST (see Table Legal input/output options used by TQGIO and TQCIO).
As of version 3.2.0 of ChemApp, TQSHOW also outputs all current system units if one or more differ from their default values (see Table Default values and units). Likewise, all target variable limits are output if any of them has been changed from their default values (see Table Options for modifying upper and lower limits of target variables using TQCLIM). TQSHOW also adds a list of all phases and phase constituents that are currently set DORMANT or ELIMINATED (see Changing the status of phases and constituents).
- See
- TQSTTP
5.9 TQGETR
GET-RESULT
Use TQGETR to get calculation results obtained by the last call to TQCE, TQCEL, TQMAP, or TQMAPL.
Synopsis
- FORTRAN:
- CALL TQGETR(OPTION,INDEXP,INDEX,VAL,NOERR)
- C:
- tqgetr(option,indexp,index,&val,&noerr);
- Pascal:
- tqgetr(option,indexp,index,val,noerr);
- Basic:
- Call tqgetr(option,indexp,index,val,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table State variables for which results can be obtained with the subroutine TQGETR |
| INDEXP | INTEGER | Set to the index number for a phase, if necessary |
| INDEX | INTEGER | Set to the index number for a constituent or component, if necessary |
| VAL | DOUBLE PRECISION or array of DOUBLE PRECISION | Returns the value(s) for the state variable |
| NOERR | INTEGER | Returns an error number |
OPTION is character input identifying the variable for which the calculated results are to be returned (see Table State variables for which results can be obtained with the subroutine TQGETR ). The integers INDEXP and INDEX are used to denote if a single value is expected on return or multiple values (see Table Definition of the indices INDEXP and INDEX in TQGETR). In the first case, VAL need only be a variable of type DOUBLE. In the latter case, VAL has to be an array of appropriate size of DOUBLE PRECISION values. VAL is returned with the default unit, or a unit set using TQCSU.
Data for any phase that might de-mix (see also Data-files with preset suppression of species) is entered twice in a ChemApp thermodynamic data-file and ChemApp automatically adds '#1' and '#2' to its name. If results are wanted for a specific composition of the two mixtures, '#1' indicates the duplicated phase with the higher activity. Also note that within a miscibility gap, '#1' denotes the duplicated phase with the higher amount.
- See also
- TQGDPC
! Retrieving results from equilibrium calculations PROGRAM CAF19 IMPLICIT NONE INTEGER NOERR, ISIO2, ICO2, INDEXP, NSC DOUBLE PRECISION TP(2), VALS(2), VALUE, VALUES(3) CHARACTER SCNAME*24 ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Define a sample input stream ('INPUTS') ! Use the array called TP to pass the information on temperature and ! pressure (298.15 K and 1 bar) of the stream to TQSTTP TP(1) = 298.15D0 TP(2) = 1.D0 CALL TQSTTP('INPUTS ', TP, NOERR) ! Define the amounts of constituents of this stream (1 mol quartz and ! 0.5 mol CO2) CALL TQINP('SiO2(quartz)', ISIO2, NOERR) CALL TQSTCA('INPUTS ', ISIO2, 0, 1.D0, NOERR) CALL TQINPC('CO2 ', 1, ICO2, NOERR) CALL TQSTCA('INPUTS ', 1, ICO2, 0.5D0, NOERR) ! Set the temperature at which the two streams should react CALL TQSTEC('T ', 0, 2500.D0, NOERR) ! Calculate the equilibrium CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 2500 K
P = 1 bar
V = 111.19 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/K PRESSURE/bar STREAM
SiO2(quartz) 1.0000E+00 298.15 1.0000E+00 1
CO2/GAS/ 5.0000E-01 298.15 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
CO2 4.3364E-01 8.1066E-01 8.1066E-01
CO 6.6363E-02 1.2406E-01 1.2406E-01
O2 3.2435E-02 6.0635E-02 6.0635E-02
O 1.9270E-03 3.6023E-03 3.6023E-03
SiO 4.3397E-04 8.1128E-04 8.1128E-04
SiO2 1.2540E-04 2.3442E-04 2.3442E-04
O3 T 2.4872E-09 4.6497E-09 4.6497E-09
Si 1.1737E-11 2.1942E-11 2.1942E-11
C 6.4393E-15 1.2038E-14 1.2038E-14
Si2 1.6425E-21 3.0705E-21 3.0705E-21
SiC 6.2162E-22 1.1621E-21 1.1621E-21
C2 1.1943E-22 2.2326E-22 2.2326E-22
Si2C 2.4986E-27 4.6711E-27 4.6711E-27
C3 6.7723E-29 1.2660E-28 1.2660E-28
Si3 1.2802E-30 2.3933E-30 2.3933E-30
TOTAL: 5.3492E-01 1.0000E+00 1.0000E+00
mol ACTIVITY
SiO2(liquid) 9.9944E-01 1.0000E+00
SiO2(cristobali) 0.0000E+00 8.9012E-01
SiO2(tridymite) T 0.0000E+00 8.5795E-01
SiO2(quartz) T 0.0000E+00 7.4840E-01
C 0.0000E+00 7.1792E-08
Si 0.0000E+00 4.7391E-09
SiC 0.0000E+00 1.5332E-15
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
1.23361E+02 2.53297E+05 2.13883E+02 -6.08043E+05 9.87954E+01
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.86724E+02 -8.54155E+05 3.62228E+02 -1.75972E+06 1.11190E+02
Mole fraction of system components:
GAS
C 0.33296
O 0.66667
Si 3.7249E-04
Data on 3 constituents marked with 'T' are extrapolated outside their valid
temperature range
50 FORMAT(1X,A,G12.5,A) ! A number of the results retrieved below can also be found in the ! ChemSage result table output above by TQCEL. If you do not want a ! ChemSage result table, for instance because your application program ! has a graphical user interface and cannot make use of the table, call ! TQCE instead to calculate the equilibrium. ! Get the pressure, which is 1 bar, since this is the default. CALL TQGETR('P ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Equilibrium pressure: ', VALUE, ' bar'
Equilibrium pressure: 1.0000 bar
! Get the calculated _total_ equilibrium volume CALL TQGETR('VT ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Equilibrium volume (total): ', VALUE, ' dm^3'
Equilibrium volume (total): 111.19 dm^3
! Get the calculated _change_ in volume. Note that options 'V' and 'VT' ! _only_ give different values if streams are used _and_ the whole ! system is selected, in which case 'V' retrieves the _change_ in volume ! between the reaction products and the stream constituents. In all ! other cases, both 'VT' and 'V' give the _total_ volume. CALL TQGETR('V ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Change in volume: ', VALUE, ' dm^3'
Change in volume: 98.795 dm^3
! Get the temperature, which is 2500 K since we set this particular ! condition for the equilibrium calculation CALL TQGETR('T ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Equilibrium temperature: ', VALUE, ' K'
Equilibrium temperature: 2500.0 K
! Get the equilibrium amount of liquid SiO2 CALL TQINP('SiO2(liquid)', INDEXP, NOERR) CALL TQGETR('A ', INDEXP, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Equilibrium amount of SiO2(liquid): ', * VALUE, ' mol'
Equilibrium amount of SiO2(liquid): 0.99944 mol
! Get the incoming amount of CO2/GAS/ that was input for the last ! equilibrium calculation CALL TQGETR('IA ', 1, ICO2, VALUE, NOERR) WRITE(*,FMT=50) 'Incoming amount of CO2/GAS/: ', * VALUE, ' mol'
Incoming amount of CO2/GAS/: 0.50000 mol
! Get the chemical potential of SiO2(quartz) CALL TQGETR('MU ', 1, ISIO2, VALUE, NOERR) WRITE(*,FMT=50) 'Chemical potential of SiO2(quartz): ', * VALUE, ' J/mol'
Chemical potential of SiO2(quartz): -4363.3 J/mol
! Get the activity of SiO2(quartz) CALL TQGETR('AC ', 1, ISIO2, VALUE, NOERR) WRITE(*,FMT=50) 'Activity of SiO2(quartz): ', * VALUE, ' '
Activity of SiO2(quartz): 0.81066
! Get the activity of the first system component (carbon). Note that the ! activities of system components are given in absolute values, whereas ! relative values are obtained for phases and constituents. CALL TQGETR('AC ', 0, 1, VALUE, NOERR) WRITE(*,FMT=50) 'Activity of system component C: ', * VALUE, ' '
Activity of system component C: 0.27651E-08
! Get the changes in the extensive property values between the reaction ! products and the reactants given as the stream constituents in the ! current units for the whole system (note that these are relative ! values) WRITE(*,'(A)') ' Changes in extensive properties:' CALL TQGETR('CP ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Delta CP: ', VALUE, ' J/K' CALL TQGETR('H ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Delta H: ', VALUE, ' J' CALL TQGETR('S ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Delta S: ', VALUE, ' J/K' CALL TQGETR('G ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Delta G: ', VALUE, ' J' CALL TQGETR('V ', 0, 0, VALUE, NOERR) WRITE(*,FMT=50) 'Delta V: ', VALUE, ' dm^3'
Changes in extensive properties: Delta CP: 123.36 J/K Delta H: 0.25330E+06 J Delta S: 213.88 J/K Delta G: -0.60804E+06 J Delta V: 98.795 dm^3
! Get the Gibbs energy value for CO2/GAS/ CALL TQGETR('G ', 1, ICO2, VALUE, NOERR) WRITE(*,FMT=50) 'G(CO2/GAS/): ', VALUE, ' J'
G(CO2/GAS/): -0.46935E+06 J
! Get the Gibbs energy value for CO2/GAS/ per amount unit CALL TQGETR('GM ', 1, ICO2, VALUE, NOERR) WRITE(*,FMT=50) 'G_M(CO2/GAS/): ', VALUE, ' J/mol'
G_M(CO2/GAS/): -0.10824E+07 J/mol
! Get the mole fraction of system component C in the system CALL TQGETR('X ', 0, 1, VALUE, NOERR) WRITE(*,FMT=50) 'Mole fraction of C in the system: ', * VALUE, ' '
Mole fraction of C in the system: 0.11111
! Get the mole fraction of system component C in the gas phase CALL TQGETR('XP ', 1, 1, VALUE, NOERR) WRITE(*,FMT=50) 'Mole fraction of C in the gas phase: ', * VALUE, ' '
Mole fraction of C in the gas phase: 0.33296
! Now get the total amount of system component C in the gas phase CALL TQGETR('AP ', 1, 1, VALUE, NOERR) WRITE(*,FMT=50) 'Amount of system component C' // * ' in the GAS phase: ', VALUE, ' mol'
Amount of system component C in the GAS phase: 0.50000 mol
! Now get the total amount of all system components in the gas phase, ! having TQGETR return all three values in one array CALL TQGETR('AP ', 1, -1, VALUES, NOERR) WRITE(*,'(A)') ' Amounts of all system components' // * ' in the GAS phase:' DO NSC = 1, 3 CALL TQGNSC(NSC, SCNAME, NOERR) WRITE(*,FMT=50) 'Amount of ' // SCNAME // * ' in the GAS phase: ', VALUES(NSC), ' mol' ENDDO END
Amounts of all system components in the GAS phase: Amount of C in the GAS phase: 0.50000 mol Amount of O in the GAS phase: 1.0011 mol Amount of Si in the GAS phase: 0.55936E-03 mol
/* Program cac19 */ /* Retrieving results from equilibrium calculations */ #include "cacint.h" int main() { LI noerr, iSiO2, iCO2, indexp, nsc; DB TP[2], vals[2], value, values[3]; char scname[TQSTRLEN]; /* Initialise ChemApp */ tqini(&noerr); /* Open data-file for reading */ tqopna("cosi.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Define a sample input stream ("INPUTS") */ /* Use the array called TP to pass the information on temperature and pressure (298.15 K and 1 bar) of the stream to tqsttp */ TP[0] = 298.15 ; TP[1] = 1.0; tqsttp("INPUTS", TP, &noerr); /* Define the amounts of constituents of this stream (1 mol quartz and 0.5 mol CO2) */ tqinp("SiO2(quartz)", &iSiO2, &noerr); tqstca("INPUTS", iSiO2, 0, 1.0, &noerr); tqinpc("CO2", 1, &iCO2, &noerr); tqstca("INPUTS", 1, iCO2, 0.5, &noerr); /* Set the temperature at which the two streams should react */ tqstec("T", 0, 2500.0, &noerr); /* Calculate the equilibrium */ tqcel(" ", 0, 0, vals, &noerr);
T = 2500 K
P = 1 bar
V = 111.19 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/K PRESSURE/bar STREAM
SiO2(quartz) 1.0000E+00 298.15 1.0000E+00 1
CO2/GAS/ 5.0000E-01 298.15 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
CO2 4.3364E-01 8.1066E-01 8.1066E-01
CO 6.6363E-02 1.2406E-01 1.2406E-01
O2 3.2435E-02 6.0635E-02 6.0635E-02
O 1.9270E-03 3.6023E-03 3.6023E-03
SiO 4.3397E-04 8.1128E-04 8.1128E-04
SiO2 1.2540E-04 2.3442E-04 2.3442E-04
O3 T 2.4872E-09 4.6497E-09 4.6497E-09
Si 1.1737E-11 2.1942E-11 2.1942E-11
C 6.4393E-15 1.2038E-14 1.2038E-14
Si2 1.6425E-21 3.0705E-21 3.0705E-21
SiC 6.2162E-22 1.1621E-21 1.1621E-21
C2 1.1943E-22 2.2326E-22 2.2326E-22
Si2C 2.4986E-27 4.6711E-27 4.6711E-27
C3 6.7723E-29 1.2660E-28 1.2660E-28
Si3 1.2802E-30 2.3933E-30 2.3933E-30
TOTAL: 5.3492E-01 1.0000E+00 1.0000E+00
mol ACTIVITY
SiO2(liquid) 9.9944E-01 1.0000E+00
SiO2(cristobali) 0.0000E+00 8.9012E-01
SiO2(tridymite) T 0.0000E+00 8.5795E-01
SiO2(quartz) T 0.0000E+00 7.4840E-01
C 0.0000E+00 7.1792E-08
Si 0.0000E+00 4.7391E-09
SiC 0.0000E+00 1.5332E-15
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
1.23361E+02 2.53297E+05 2.13883E+02 -6.08043E+05 9.87954E+01
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.86724E+02 -8.54155E+05 3.62228E+02 -1.75972E+06 1.11190E+02
Mole fraction of system components:
GAS
C 0.33296
O 0.66667
Si 3.7249E-04
Data on 3 constituents marked with 'T' are extrapolated outside their valid
temperature range
/* A number of the results retrieved below can also be found in the ChemSage result table output above by tqcel. If you do not want a ChemSage result table, for instance because your application program has a graphical user interface and cannot make use of the table, call tqce instead to calculate the equilibrium. */ /* Get the pressure, which is 1 bar, since this is the default. */ tqgetr("P", 0, 0, &value, &noerr); printf("Equilibrium pressure: %g bar\n", value);
Equilibrium pressure: 1 bar
/* Get the calculated equilibrium volume */ tqgetr("VT", 0, 0, &value, &noerr); printf("Equilibrium volume (total): %g dm^3\n", value);
Equilibrium volume (total): 111.19 dm^3
/* Get the calculated _change_ in volume. Note that options "V" and "VT" _only_ give different values if streams are used _and_ the whole system is selected, in which case "V" retrieves the _change_ in volume between the reaction products and the stream constituents. In all other cases, both "VT" and "V" give the _total_ volume. */ tqgetr("V", 0, 0, &value, &noerr); printf("Change in volume: %g dm^3\n", value);
Change in volume: 98.7954 dm^3
/* Get the temperature, which is 2500 K, since we set this particular condition for the equilibrium calculation */ tqgetr("T", 0, 0, &value, &noerr); printf("Equilibrium temperature: %g K\n", value);
Equilibrium temperature: 2500 K
/* Get the equilibrium amount of liquid SiO2 */ tqinp("SiO2(liquid)", &indexp, &noerr); tqgetr("a", indexp, 0, &value, &noerr); printf("Equilibrium amount of SiO2(liquid): %g mol\n", value);
Equilibrium amount of SiO2(liquid): 0.999441 mol
/* Get the incoming amount of CO2/GAS/ that was input for the last equilibrium calculation */ tqgetr("ia", 1, iCO2, &value, &noerr); printf("Incoming amount of CO2/GAS/: %g mol\n", value);
Incoming amount of CO2/GAS/: 0.5 mol
/* Get the chemical potential of SiO2(quartz) */ tqgetr("mu", 1, iSiO2, &value, &noerr); printf("Chemical potential of SiO2(quartz): %g J/mol\n", value);
Chemical potential of SiO2(quartz): -4363.26 J/mol
/* Get the activity of SiO2(quartz) */ tqgetr("ac", 1, iSiO2, &value, &noerr); printf("Activity of SiO2(quartz): %g\n", value);
Activity of SiO2(quartz): 0.810657
/* Get the activity of the first system component (carbon). Note that the activities of system components are given in absolute values, whereas relative values are obtained for phases and constituents. */ tqgetr("ac", 0, 1, &value, &noerr); printf("Activity of system component C: %g\n", value);
Activity of system component C: 2.76514e-09
/* Get the changes in the extensive property values between the reaction products and the reactants given as the stream constituents in the current units for the whole system (note that these are relative values) */ printf("Changes in extensive properties:\n"); tqgetr("CP", 0, 0, &value, &noerr); printf("Delta CP: %g J/K\n", value); tqgetr("H", 0, 0, &value, &noerr); printf("Delta H: %g J\n", value); tqgetr("S", 0, 0, &value, &noerr); printf("Delta S: %g J/K\n", value); tqgetr("G", 0, 0, &value, &noerr); printf("Delta G: %g J\n", value); tqgetr("V", 0, 0, &value, &noerr); printf("Delta V: %g dm^3\n", value);
Changes in extensive properties: Delta CP: 123.361 J/K Delta H: 253297 J Delta S: 213.883 J/K Delta G: -608043 J Delta V: 98.7954 dm^3
/* Get the Gibbs energy value for CO2/GAS/ */ tqgetr("G", 1, iCO2, &value, &noerr); printf("G(CO2/GAS/): %g J\n", value);
G(CO2/GAS/): -469351 J
/* Get the Gibbs energy value for CO2/GAS/ per amount unit */ tqgetr("GM", 1, iCO2, &value, &noerr); printf("G_M(CO2/GAS/): %g J/mol\n", value);
G_M(CO2/GAS/): -1.08236e+06 J/mol
/* Get the mole fraction of system component C in the system */ tqgetr("X", 0, 1, &value, &noerr); printf("Mole fraction of C in the system: %g\n", value);
Mole fraction of C in the system: 0.111111
/* Get the mole fraction of system component C in the gas phase */ tqgetr("XP", 1, 1, &value, &noerr); printf("Mole fraction of C in the gas phase: %g\n", value);
Mole fraction of C in the gas phase: 0.332961
/* Now get the total amount of all system components in the gas phase, having tqgetr return all three values in one array */ tqgetr("ap", 1, -1, values, &noerr); printf("Amounts of all system components in the GAS phase:\n"); for (nsc = 0; nsc < 3; nsc++) { tqgnsc(nsc+1, scname, &noerr); printf("Amount of %s in the GAS phase: %g mol\n", scname, values[nsc]); } return 0; }
Amounts of all system components in the GAS phase: Amount of C in the GAS phase: 0.5 mol Amount of O in the GAS phase: 1.00112 mol Amount of Si in the GAS phase: 0.000559364 mol
5.10 TQGDPC
GET-THERMODYNAMIC-DATA-OF-PHASE-CONSTITUENT
Use TQGDPC to get thermodynamic data for a phase constituent.
Synopsis
- FORTRAN:
- CALL TQGDPC(OPTION,INDEXP,INDEXC,VAL,NOERR)
- C:
- tqgdpc(option,indexp,indexc,&val,&noerr);
- Pascal:
- tqgdpc(option,indexp,indexc,val,noerr);
- Basic:
- Call tqgdpc(option,indexp,indexc,val,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| OPTION | CHARACTER | Set to a string as shown in Table Thermodynamic quantities that can be calculated using TQGDPC |
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXC | INTEGER | Set to the index number for a constituent |
| VAL | DOUBLE PRECISION | Returns the value of the thermodynamic quantity |
| NOERR | INTEGER | Returns an error number |
OPTION is character input identifying the thermodynamic quantity (see Table Thermodynamic quantities that can be calculated using TQGDPC). VAL (for a single constituent, selected using INDEXP and INDEXC) is returned dimensionless and is calculated for the default temperature, or for the last temperature set using TQSETC or TQSTEC, or calculated using TQCE/TQMAP.
Note: In order to obtain quantities having dimensions, VAL needs to be multiplied by R*T (gas constant times temperature). See the code below for an example.
! Calculate thermodynamic quantities PROGRAM CAF20 IMPLICIT NONE INTEGER NOERR, NUMCON, ISIO2 DOUBLE PRECISION VALUE, R, T ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) 50 FORMAT(1X,A,G12.5,A) ! Defining the gas constant in J/mol.K R=8.31451 ! Get thermochemical function values for SiO2(quartz) at 500 K T = 500.D0 CALL TQSETC('T ', 0, 0, T, NUMCON, NOERR) CALL TQINP('SiO2(quartz)', ISIO2, NOERR) ! Note that when retrieving CP, H, S, or G, the values are returned ! dimensionless, which means they might have to be multiplied by ! R*T. Since the default amount unit is mol, results are returned for 1 ! mol. Note also that care has to be taken if a temperature unit ! different from Kelvin has been used. CALL TQGDPC('CP ', ISIO2, 0, VALUE, NOERR) WRITE(*,FMT=50) 'CP of SiO2(quartz) at 500 K: ', * VALUE*R*T, ' J/mol.K' CALL TQGDPC('H ', ISIO2, 0, VALUE, NOERR) WRITE(*,FMT=50) 'H of SiO2(quartz) at 500 K: ', * VALUE*R*T, ' J/mol'
CP of SiO2(quartz) at 500 K: 59.498 J/mol.K H of SiO2(quartz) at 500 K: -0.90000E+06 J/mol
! The option 'T' serves a special purpose: If used, it returns the end ! temperature of the current interval of the CP or G function for the ! selected phase or phase constituent. Looking at the data-file cosi.dat ! (see Appendix ref(cosidat)), one sees from the entry for SiO2(quartz) ! that for a temperature of 500 K, the temperature range of the G ! function in question extends to 848 K. CALL TQGDPC('T ', ISIO2, 0, VALUE, NOERR) WRITE(*,FMT='(A)') ' For SiO2(quartz) and T = 500 K, the present' WRITE(*,FMT=50) 'temperature interval ends at: ', VALUE, ' K'
For SiO2(quartz) and T = 500 K, the present temperature interval ends at: 848.00 K
! Setting a different temperature for which a different G function ! interval applies T = 1000.D0 CALL TQSETC('T ', 0, 0, T, NUMCON, NOERR) CALL TQGDPC('T ', ISIO2, 0, VALUE, NOERR) WRITE(*,FMT='(A)') ' For SiO2(quartz) and T = 1000 K, the present' WRITE(*,FMT=50) 'temperature interval ends at: ', VALUE, ' K'
For SiO2(quartz) and T = 1000 K, the present temperature interval ends at: 1744.2 K
! The option 'T' of TQGDPC is especially useful when the thermodynamic ! values are calculated as a function of temperature, and the ranges of ! the CP or G functions need to be known explicitly in order to locate ! transition temperatures between different modifications and determine ! the associated changes in the thermodynamic values. END
/* Program cac20 */ /* Calculate thermodynamic quantities */ #include "cacint.h" int main() { LI noerr, numcon, iSiO2; DB value, R, T; /* Initialise ChemApp */ tqini(&noerr); /* Open data-file for reading */ tqopna("cosi.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10,&noerr); /* Defining the gas constant in J/mol.K */ R = 8.31451; /* Get thermochemical function values for SiO2(quartz) at 500 K */ T = 500.0; tqsetc("T", 0, 0, T, &numcon, &noerr); tqinp("SiO2(quartz)", &iSiO2, &noerr); /* Note that when retrieving CP, H, S, or G, the values are returned dimensionless, which means they might have to be multiplied with R*T. Since the default amount unit is mol, results are returned for 1 mol. Note also that care has to be taken if a temperature unit different from Kelvin has been used. */ tqgdpc("CP", iSiO2, 0, &value, &noerr); printf("CP of SiO2(quartz) at 500 K: %g J/mol.K\n", value*R*T); tqgdpc("H", iSiO2, 0, &value, &noerr); printf("H of SiO2(quartz) at 500 K: %g J/mol\n", value*R*T);
CP of SiO2(quartz) at 500 K: 59.498 J/mol.K H of SiO2(quartz) at 500 K: -899999 J/mol
/* The option "T" serves a special purpose: If used, it returns the end temperature of the current interval of the CP or G function for the selected phase or phase constituent. Looking at the data-file cosi.dat (see Appendix ref(cosidat)), one sees from the entry for SiO2(quartz) that for a temperature of 500 K, the temperature range of the G function in question extends to 848 K. */ tqgdpc("T", iSiO2, 0, &value, &noerr); printf("For SiO2(quartz) and T = 500 K, the present\n" "temperature interval ends at: %g K\n", value);
For SiO2(quartz) and T = 500 K, the present temperature interval ends at: 848 K
/* Setting a different temperature for which a different G function interval applies */ T = 1000.0; tqsetc("T", 0, 0, T, &numcon, &noerr); tqgdpc("T", iSiO2, 0, &value, &noerr); printf("For SiO2(quartz) and T = 1000 K, the present\n" "temperature interval ends at: %g K\n", value);
For SiO2(quartz) and T = 1000 K, the present temperature interval ends at: 1744.25 K
/* The option "T" of tqgdpc is especially useful when the thermodynamic values are calculated as a function of temperature, and the ranges of the CP or G functions need to be known explicitly in order to locate transition temperatures between different modifications and determine the associated changes in the thermodynamic values. */ return 0; }
5.11 TQSTXP
GET-THERMODYNAMIC-PROPERTY-OF-A-STREAM
Use TQSTXP to get the calculated thermodynamic properties of a stream.
Important
When using data-files that contains phases described with the SUBG model (such as ASlag-liq from the FACT solution database), please make sure you read Extensive properties of streams and the SUBG model.
Added for ChemApp version 1.1.3
Synopsis
- FORTRAN:
- CALL TQSTXP(IDENTS,OPTION,VAL,NOERR)
- C:
- tqstxp(idents,option,&val,&noerr);
- Pascal:
- tqstxp(idents,option,val,noerr);
- Basic:
- Call tqstxp(idents,option,val,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| IDENTS | CHARACTER | Set to the name of a stream |
| OPTION | CHARACTER | Set to a string as shown in Table Thermodynamic quantities that can be calculated using TQSTXP |
| VAL | DOUBLE PRECISION | Returns the value of the thermodynamic quantity |
| NOERR | INTEGER | Returns an error number |
IDENTS and OPTION are both input. IDENTS is a name previously defined by calling TQSTTP. OPTION is character input identifying the thermodynamic quantity (see Table Thermodynamic quantities that can be calculated using TQSTXP). The subroutine returns the value of the desired thermodynamic quantity through VAL, using the currently selected system units. See also the notes in Appendix Extensive properties of streams for more information on how the extensive properties are calculated.
For users of ChemApp versions prior to 3.2.0: Please note that the values returned by TQSTXP always refer to the last calculated equilibrium state. If TQSTXP is called before the first equilibrium calculation has been performed using TQCE or TQMAP, VAL will be zero.
As of ChemApp version 3.2.0, TQSTXP can be called to get the calculated thermodynamic properties of a stream also before an equilibrium calculation has been performed.
- See also
- TQSTCA, TQSTEC, TQSTRM, TQSTTP, Extensive properties of streams
- See
- TQSTTP
5.12 TQGTLC
GET-CALCULATED-EQUILIBRIUM-SUBLATTICE-SITE-FRACTION
Use TQGTLC to get the calculated equilibrium sublattice site fraction.
Added for ChemApp version 3.3.1
Synopsis
- FORTRAN:
- CALL TQGTLC(INDEXP,INDEXL,INDEXC,VAL,NOERR)
- C:
- tqgtlc(indexp,indexl,indexc,&val,&noerr);
- Pascal:
- tqgtlc(indexp,indexl,indexc,val,noerr);
- Basic:
- Call tqgtlc(indexp,indexl,indexc,val,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXL | INTEGER | Set to the index number for a sublattice |
| INDEXC | INTEGER | Set to the index number for a sublattice constituent |
| VAL | DOUBLE PRECISION | Returns the value of the calculated sublattice site fraction |
| NOERR | INTEGER | Returns an error number |
The input variables have to be set to index numbers which identify a sublattice constituent (INDEXC) of a sublattice (INDEXL) of a phase (INDEXP). The output variable VAL contains the calculated sublattice site fraction of the sublattice constituent with respect to the last equilibrium calculation.
All solution phases are considered to be modelled with one or several sublattices. For one-sublattice models, the mole and site fractions are equal.
! Retrieving results for sublattice constituents PROGRAM CAF27 IMPLICIT NONE INTEGER NOERR, ISLITE, NUMCON, I, INDEXP, INDEXL, * INDEXC, NPHASE, NSL, NSLCON DOUBLE PRECISION VAL, VALS(2), RESULT CHARACTER PNAME*24, MNAME*24, CNAME*24 ! Initialise ChemApp CALL TQINI(NOERR) ! Open the thermochemical data-file subl-ex.dat for reading. It contains ! an extract of the system Co-Cr-Fe: the phase SIGMA:30, which is ! modelled according to the sublattice formalism, and the BCC phase, ! described by a Redlich-Kister polynomial. Both phases are each ! included twice, to account for miscibility gaps. CALL TQOPNA('subl-ex.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Set the temperature to 1000 K CALL TQSETC('T ', 0, 0, 1000.D0, NUMCON, NOERR) ! Set the incoming amounts to 0.25 mol Co, 0.25 mol Cr, and 0.5 mol Fe CALL TQINSC('Co ', INDEXC, NOERR) CALL TQSETC('IA ', 0, INDEXC, 0.25D0, NUMCON, NOERR) CALL TQINSC('Cr ', INDEXC, NOERR) CALL TQSETC('IA ', 0, INDEXC, 0.25D0, NUMCON, NOERR) CALL TQINSC('Fe ', INDEXC, NOERR) CALL TQSETC('IA ', 0, INDEXC, 0.50D0, NUMCON, NOERR) ! Calculate the equilibrium CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 1000 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
Co 2.5000E-01
Cr 2.5000E-01
Fe 5.0000E-01
EQUIL AMOUNT MOLE FRACTION ACTIVITY
PHASE: BCC#1 mol
Co 2.0130E-01 2.5280E-01 1.4418E-01
Cr 1.4767E-01 1.8544E-01 4.3552E-01
Fe 4.4733E-01 5.6176E-01 5.6476E-01
TOTAL: 7.9630E-01 1.0000E+00 1.0000E+00
PHASE: BCC#2 mol MOLE FRACTION ACTIVITY
Co 0.0000E+00 2.5280E-01 1.4418E-01
Cr 0.0000E+00 1.8544E-01 4.3552E-01
Fe 0.0000E+00 5.6176E-01 5.6476E-01
TOTAL: 0.0000E+00 1.0000E+00 1.0000E+00
PHASE: SIGMA:30#1 mol MOLE FRACTION ACTIVITY
Co:Cr:Co 1.2071E-02 5.9260E-02 1.1301E-01
Co:Cr:Cr 7.6446E-02 3.7528E-01 8.0896E-01
Co:Cr:Fe 2.1966E-02 1.0784E-01 2.7700E-01
Fe:Cr:Co 1.0185E-02 5.0000E-02 6.0745E-01
Fe:Cr:Cr 6.4500E-02 3.1664E-01 5.0651E-01
Fe:Cr:Fe 1.8534E-02 9.0985E-02 4.2173E-01
TOTAL: 2.0370E-01 1.0000E+00 1.0000E+00
PHASE: SIGMA:30#2 mol MOLE FRACTION ACTIVITY
Co:Cr:Co 0.0000E+00 5.9260E-02 1.1301E-01
Co:Cr:Cr 0.0000E+00 3.7528E-01 8.0896E-01
Co:Cr:Fe 0.0000E+00 1.0784E-01 2.7700E-01
Fe:Cr:Co 0.0000E+00 5.0000E-02 6.0745E-01
Fe:Cr:Cr 0.0000E+00 3.1664E-01 5.0651E-01
Fe:Cr:Fe 0.0000E+00 9.0985E-02 4.2173E-01
TOTAL: 0.0000E+00 1.0000E+00 1.0000E+00
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.26615E+01 -2.13200E+02 7.05477E+00 -7.26797E+03 0.00000E+00
Site fraction of sublattice constituents in SIGMA:30#1:
Co 0.54238 Stoichiometry = 0.33333
Fe 0.45762
---------------------------------------------------------------
Cr 1.0000 Stoichiometry = 0.13333
---------------------------------------------------------------
Co 0.10926 Stoichiometry = 0.53333
Cr 0.69192
Fe 0.19882
Mole fraction of system components:
SIGMA:30#1
Co 0.23906
Cr 0.50236
Fe 0.25858
! As can be seen in the output above, the standard ChemSage result table ! obtained with TQCEL contains a section with details on the stable ! sublattice phases. The following code shows how to retrieve similar ! information with ChemApp. ! Loop over all phases, and if a phase is stable and a sublattice phase, ! print information about the sublattices CALL TQNOP(NPHASE, NOERR) DO INDEXP=1, NPHASE ! Check if the phase is stable, otherwise we don't need to consider it CALL TQGETR('A ', INDEXP, 0, VAL, NOERR) IF (VAL .GT. 0.D0) THEN ! Get the name and model of the phase CALL TQGNP(INDEXP, PNAME, NOERR) CALL TQMODL(INDEXP, MNAME, NOERR) ! If the model name of the phase starts with 'SUB' we have a sublattice ! phase IF (INDEX(MNAME,'SUB') .GT. 0) then ! Print a header similar to the one in the ChemSage output table WRITE(*,*) 'Mole fraction of the sublattice ' // * 'constituents in ', PNAME ! Get the number of sublattices CALL TQNOSL(INDEXP, NSL, NOERR) ! Loop over all sublattices DO INDEXL=1, NSL write(*,*) 'Sublattice ',INDEXL,' :' ! Get the number of sublattice constituents CALL TQNOLC(INDEXP, INDEXL, NSLCON, NOERR) DO INDEXC=1, NSLCON ! Get the name of each sublattice constituent... CALL TQGNLC(INDEXP, INDEXL, INDEXC, CNAME, NOERR) ! ... and its mole fraction in the sublattice CALL TQGTLC(INDEXP, INDEXL, INDEXC, VAL, NOERR) WRITE(*,*) CNAME, VAL ENDDO ENDDO ENDIF ENDIF ENDDO END
Mole fraction of the sublattice constituents in SIGMA:30#1 Sublattice 1 : Co 0.54237736793020974 Fe 0.45762263206978993 Sublattice 2 : Cr 0.99999999999999967 Sublattice 3 : Co 0.10925936359569255 Cr 0.69191988174638075 Fe 0.19882075465792631
/* Program cac27 */ /* Retrieving results for sublattice constituents */ #include "cacint.h" int main() { LI noerr, islite, numcon, i, indexp, indexl, indexc, nphase, nsl, nslcon; DB val, vals[2], result; char pname[TQSTRLEN], mname[TQSTRLEN], cname[TQSTRLEN]; /* Initialise ChemApp */ tqini(&noerr); /* Open the thermochemical data-file subl-ex.dat for reading. It contains an extract of the system Co-Cr-Fe: the phase SIGMA:30, which is modelled according to the sublattice formalism, and the BCC phase, described by a Redlich-Kister polynomial. Both phases are each included twice, to account for miscibility gaps. */ tqopna("subl-ex.dat",10,&noerr); /* Read data-file */ tqrfil(&noerr); /* Close data-file */ tqclos(10, &noerr); /* Set the temperature to 1000 K */ tqsetc("T", 0, 0, 1000.0, &numcon, &noerr); /* Set the incoming amounts to 0.25 mol Co, 0.25 mol Cr, and 0.5 mol Fe */ tqinsc("Co", &indexc, &noerr); tqsetc("ia", 0, indexc, 0.25, &numcon, &noerr); tqinsc("Cr", &indexc, &noerr); tqsetc("ia", 0, indexc, 0.25, &numcon, &noerr); tqinsc("Fe", &indexc, &noerr); tqsetc("ia", 0, indexc, 0.50, &numcon, &noerr); /* Calculate the equilibrium */ tqcel(" ", 0, 0, vals, &noerr);
T = 1000 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
Co 2.5000E-01
Cr 2.5000E-01
Fe 5.0000E-01
EQUIL AMOUNT MOLE FRACTION ACTIVITY
PHASE: BCC#1 mol
Co 2.0130E-01 2.5280E-01 1.4418E-01
Cr 1.4767E-01 1.8544E-01 4.3552E-01
Fe 4.4733E-01 5.6176E-01 5.6476E-01
TOTAL: 7.9630E-01 1.0000E+00 1.0000E+00
PHASE: BCC#2 mol MOLE FRACTION ACTIVITY
Co 0.0000E+00 2.5280E-01 1.4418E-01
Cr 0.0000E+00 1.8544E-01 4.3552E-01
Fe 0.0000E+00 5.6176E-01 5.6476E-01
TOTAL: 0.0000E+00 1.0000E+00 1.0000E+00
PHASE: SIGMA:30#1 mol MOLE FRACTION ACTIVITY
Co:Cr:Co 1.2071E-02 5.9260E-02 1.1301E-01
Co:Cr:Cr 7.6446E-02 3.7528E-01 8.0896E-01
Co:Cr:Fe 2.1966E-02 1.0784E-01 2.7700E-01
Fe:Cr:Co 1.0185E-02 5.0000E-02 6.0745E-01
Fe:Cr:Cr 6.4500E-02 3.1664E-01 5.0651E-01
Fe:Cr:Fe 1.8534E-02 9.0985E-02 4.2173E-01
TOTAL: 2.0370E-01 1.0000E+00 1.0000E+00
PHASE: SIGMA:30#2 mol MOLE FRACTION ACTIVITY
Co:Cr:Co 0.0000E+00 5.9260E-02 1.1301E-01
Co:Cr:Cr 0.0000E+00 3.7528E-01 8.0896E-01
Co:Cr:Fe 0.0000E+00 1.0784E-01 2.7700E-01
Fe:Cr:Co 0.0000E+00 5.0000E-02 6.0745E-01
Fe:Cr:Cr 0.0000E+00 3.1664E-01 5.0651E-01
Fe:Cr:Fe 0.0000E+00 9.0985E-02 4.2173E-01
TOTAL: 0.0000E+00 1.0000E+00 1.0000E+00
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.26615E+01 -2.13200E+02 7.05477E+00 -7.26797E+03 0.00000E+00
Site fraction of sublattice constituents in SIGMA:30#1:
Co 0.54238 Stoichiometry = 0.33333
Fe 0.45762
---------------------------------------------------------------
Cr 1.0000 Stoichiometry = 0.13333
---------------------------------------------------------------
Co 0.10926 Stoichiometry = 0.53333
Cr 0.69192
Fe 0.19882
Mole fraction of system components:
SIGMA:30#1
Co 0.23906
Cr 0.50236
Fe 0.25858
/* As can be seen in the output above, the standard ChemSage result table obtained with TQCEL contains a section with details on the stable sublattice phases. The following code shows how to retrieve similar information with ChemApp. */ /* Loop over all phases, and if a phase is stable and a sublattice phase, print information about the sublattices */ tqnop(&nphase, &noerr); for (indexp = 1; indexp<=nphase; indexp++) { /* Check if the phase is stable, otherwise we don't need to consider it */ tqgetr("a", indexp, 0, &val, &noerr); if (val > 0) { /* Get the name and model of the phase */ tqgnp(indexp, pname, &noerr); tqmodl(indexp, mname, &noerr); /* If the model name of the phase starts with 'SUB' we have a sublattice phase */ if (strncmp(mname, "SUB", 3) == 0) { /* Print a header similar to the one in the ChemSage output table */ printf("Mole fraction of the sublattice constituents in %s\n", pname); /* Get the number of sublattices */ tqnosl(indexp, &nsl, &noerr); /* Loop over all sublattices */ for (indexl=1; indexl<=nsl; indexl++) { printf("Sublattice %li\n", indexl); /* Get the number of sublattice constituents */ tqnolc(indexp, indexl, &nslcon, &noerr); for (indexc=1; indexc<=nslcon; indexc++) { /* Get the name of each sublattice constituent... */ tqgnlc(indexp, indexl, indexc, cname, &noerr); /* ... and its mole fraction in the sublattice */ tqgtlc(indexp, indexl, indexc, &val, &noerr); printf("%-25s %f\n", cname, val); } } } } } return 0; }
Mole fraction of the sublattice constituents in SIGMA:30#1 Sublattice 1 Co 0.542377 Fe 0.457623 Sublattice 2 Cr 1.000000 Sublattice 3 Co 0.109259 Cr 0.691920 Fe 0.198821
5.13 TQBOND
GET-CALCULATED-QUADRUPLET-OR-PAIR-FRACTION
Use TQBOND to get a calculated quadruplet or pair fraction (only for the models SUBG, QUAS and QSOL, see Table Identifiers of phase model names as returned by TQMODL).
Added for ChemApp version 5.4.1
Synopsis
- FORTRAN:
- CALL TQBOND(INDEXP,INDEXA,INDEXB,INDEXC,INDEXD,VAL,NOERR)
- C:
- tqbond(indexp,indexa,indexb,indexc,indexd,&val,&noerr);
- Pascal:
- tqbond(indexp,indexa,indexb,indexc,indexd,val,noerr);
- Basic:
- Call tqbond(indexp,indexa,indexb,indexc,indexd,val,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXA | INTEGER | Input (see description below) |
| INDEXB | INTEGER | Input (see description below) |
| INDEXC | INTEGER | Input (see description below) |
| INDEXD | INTEGER | Input (see description below) |
| VAL | DOUBLE PRECISION | Returns the quadruplet or pair fraction |
| NOERR | INTEGER | Returns an error number |
INDEXP, INDEXA, INDEXB, INDEXC and INDEXD are all integer input. The output variable VAL is of type DOUBLE PRECISION and contains the fraction. The four indices INDEXA - INDEXD are used for model SUBG and denote index numbers for sublattice constituents. Note that an index number of a constituent of the second sublattice = (total number of constituents of the first sublattice + index number within the second sublattice). The index numbers may be entered in any order, but two sublattice constituents must belong to the first sublattice and two to the second. INDEXA and INDEXB are entered for models QUAS and QSOL and contain index numbers for phase constituents entered in any order.
5.14 TQERR
GET-ERROR-MESSAGE
Use TQERR to get an error message.
Synopsis
- FORTRAN:
- CALL TQERR(MESS,NOERR)
- C:
- tqerr(&mess,&noerr);
- Pascal:
- tqerr(mess,noerr);
- Basic:
- Call tqerr(mess,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| MESS | Vector (1-dim. array) of CHARACTER | Returns an error message |
| NOERR | INTEGER | Returns an error number |
All ChemApp error messages are written sequentially to the unit/file associated with ERROR (see Table Legal input/output options used by TQGIO and TQCIO and TQCIO on how to change the associated unit/file and on how to prevent the automatic output of error messages).
Alternatively, they can also be retrieved individually after each ChemApp subroutine call by invoking TQERR. MESS is a CHARACTER array with 3 elements each 80 characters long. These three entries in MESS contain the error message. List of Error Messages contains a list of all possible error messages and their associated numbers.
Note that it is only necessary to call TQERR if the error variable (NOERR) returned by the most recent previous call to a ChemApp subroutine contains a positive value. Otherwise, a blank return by TQERR indicates 'no error'.
! Retrieve error messages PROGRAM CAF21 IMPLICIT NONE INTEGER NOERR, NPHASE CHARACTER MESS(3)*80 ! Initialise ChemApp CALL TQINI(NOERR) ! TQERR is especially useful if is undesirable that ChemApp writes ! error messages to the standard output unit (usually 6, the connected ! terminal). If the application program which calls ChemApp has a ! graphical user interface, ChemApp error messages written to the ! default output unit might either not be displayed at all, or might ! corrupt the graphical interface window. In these cases it is more ! useful to have ChemApp not output error messages automatically, but ! retrieve them using TQERR, if the error variable returned by a ChemApp ! subroutine indicates that an error occurred. ! Prevent the error messages from being displayed automatically CALL TQCIO('ERROR ', 0, NOERR) ! Provoke an error by trying to get the number of phases ! before even having read a thermochemical data-file CALL TQNOP(NPHASE, NOERR) IF (NOERR .NE. 0) THEN WRITE(*,FMT='(A,I4)') ' ChemApp reports error no.', NOERR ! Retrieve the error message using TQERR and the character array MESS CALL TQERR(MESS,NOERR) ! To display the message, print all three strings of MESS : WRITE(UNIT=*,FMT='(3(A,/))') MESS(1), MESS(2), MESS(3) ENDIF END
ChemApp reports error no. 104 A thermodynamic data-file must be read first The error appeared after a call of "TQNOP "
/* Program cac21 */ /* Retrieve error messages */ #include "cacint.h" int main() { LI noerr, nphase; int i; /* Initialise ChemApp */ tqini(&noerr); /* tqerr is especially useful if is undesirable that ChemApp writes error messages to standard output. If the application program which calls ChemApp has a graphical user interface, ChemApp error messages written to standard output might either not be displayed at all, or might corrupt the graphical interface window. In these cases it is more useful to have ChemApp not output error messages automatically, but retrieve them using tqerr, if the error variable returned by a ChemApp function indicates that an error occurred. */ /* Prevent the error messages from being displayed automatically */ tqcio("ERROR", 0, &noerr); /* Provoke an error by trying to get the number of phases before even having read a thermochemical data-file */ tqnop(&nphase, &noerr); if (noerr) { printf(" ChemApp reports error no. %li\n", noerr); /* Retrieve the error message using tqerr. The variable TQERRMSG to be used for that purpose is declared in cacint.c as TQERRMSG[3][80] */ tqerr(TQERRMSG,&noerr); /* To display the message, print all three strings of TQERRMSG: */ for(i=0;i<3;i++) printf("%s\n",TQERRMSG[i]); } return 0; }
ChemApp reports error no. 104 A thermodynamic data-file must be read first The error appeared after a call of "TQNOP "
6 Data Manipulation Subroutines
If the thermochemical data-file used is in ASCII format (see ASCII data-files), the subroutines listed in this chapter allow for the retrieval and manipulation of the thermochemical data in memory.
TQGDAT, TQLPAR, and TQGPAR are used to retrieve thermodynamic data, while TQCDAT is used to change thermodynamic data in memory. If the changes are to be saved permanently, TQWASC needs to be called to write the current state of all thermodynamic data in memory back to a ChemApp data-file in ASCII format.
TQCDAT should be used with caution, and only by users with a good knowledge of the structures and models applied in the thermodynamic data file that is used.
| Subroutine | Function |
|---|---|
| TQGDAT | Get selected thermodynamic data of a phase constituent |
| TQLPAR | List all excess Gibbs energy or all excess magnetic interactions of a phase |
| TQGPAR | Get selected excess Gibbs energy or excess magnetic parameters of a phase |
| TQCDAT | Change selected data of a specified phase constituent or of a specified phase |
| TQWASC | Write a thermodynamic data-file in ASCII format |
| Option | Variable |
|---|---|
| H | Enthalpy/J at 298.15 K |
| S | Entropy/J.K-1 at 298.15 K |
| Cp | Heat capacity expression/J.K-1 or Helgeson terms for the models PIHZ, HELZ, HTSZ, HTWZ or HTDZ |
| Tt | Upper temperature limit/K for a Cp range |
| Ht | Transformation enthalpy/J at the upper temperature limit for a Cp range |
| V | Molar volume data in input order of a condensed phase constituent |
| Gc | Critical properties in input order of a gas phase constituent |
| M | Curie/Neel temperature and average magnetic moment per atom for a magnetic phase constituent |
| Ch | Charge of an aqueous phase constituent |
6.1 TQGDAT
GET-INPUT-THERMODYNAMIC-DATA-OF-PHASE-CONSTITUENT
Use TQGDAT to get selected thermodynamic data of a specified phase constituent loaded from a thermodynamic data-file in ASCII format.
Synopsis
- FORTRAN:
- CALL TQGDAT(INDEXP,INDEXC,OPTION,INDEXR,NVALV,VALV,NOERR)
- C:
- tqgdat(indexp,indexc,option,indexr,&nvalv,&valv,&noerr);
- Pascal:
- tqgdat(indexp,indexc,option,indexr,nvalv,valv,noerr);
- Basic:
- Call tqgdat(indexp,indexc,option,indexr,nvalv,valv,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| INDEXC | INTEGER | Set to the index number for a constituent |
| OPTION | CHARACTER | Set to a string (see Table Options for use with TQGDAT) |
| INDEXR | INTEGER | Set to the index number of a Cp expression (used for the options Cp, Tt and Ht, for the other options INDEXR is a dummy) |
| NVALV | INTEGER | Returns the number of values in the vector VALV |
| VALV | DOUBLE PRECISION or vector (1-dim. array) of DOUBLE PRECISION | Returns NVALV values of the quantity requested |
| NOERR | INTEGER | Returns an error number |
INDEXP and INDEXC, identifying the constituent for which the data are to be returned, must both be greater than zero. OPTION is a character input identifying the quantity to be returned. The NVALV values of this quantity will be stored in the vector VALV with default units. INDEXR identifies a specific Cp range.
Note that heat capacity data will be returned also for constituents with stored values of standard Gibbs energy. For extended Cp equations with more than four terms alternating Cp terms and powers will be returned for the extended terms.
The array VALV should be declared to hold 25 elements.
! Retrieving and changing thermodynamic data PROGRAM CAF33 IMPLICIT NONE INTEGER NOERR, INDEXP, NVALV, I, J, NOPAR, NOEXPR, NVALA INTEGER LGTPAR(1999) DOUBLE PRECISION VALV(25),VALA(20,18) CHARACTER*156 CHRPAR(1999) ! Initialise ChemApp CALL TQINI(NOERR) ! Open the thermochemical data-file fec.dat (system Fe-C) ! for reading CALL TQOPNA('fec.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! NOTE: Do not yet close the data-file, otherwise tqwasc (see below) ! won't be able to access the commentary block in fec.dat in order ! to attach it to the new file. ! Get the index number of phase 'C_GRAPHITE' CALL TQINP('C_GRAPHITE ', INDEXP, NOERR) ! Retrieve Cp coefficients for the first Cp-range of C_GRAPHITE CALL TQGDAT(INDEXP, 1, 'Cp ', 1, NVALV, VALV, NOERR) ! Print coefficients DO I=1, NVALV WRITE(UNIT=*,FMT=*) 'VALV(',I,') is ', VALV(I) ENDDO
VALV( 1 ) is 24.300000000000001 VALV( 2 ) is 9.4459999999999998E-004 VALV( 3 ) is -0.0000000000000000 VALV( 4 ) is -5125200.0000000000 VALV( 5 ) is 1585800000.0000000 VALV( 6 ) is -3.0000000000000000 VALV( 7 ) is -144000000000.00000 VALV( 8 ) is -4.0000000000000000
! Get the phase index number of phase 'Fe3C_CEMENTITE' CALL TQINP('Fe3C_CEMENTITE ', INDEXP, NOERR) ! Retrieve Enthalpy/J at 298.15 K of Fe3C_CEMENTITE CALL TQGDAT(INDEXP, 1, 'H ', 1, NVALV, VALV, NOERR) WRITE(UNIT=*,FMT=*) 'Enthalpy/J at 298.15 K of Fe3C_CEMENTITE: ', * VALV(1) ! Set the enthalpy/J at 298.15 K of Fe3C_CEMENTITE to a new value CALL TQCDAT(1, 0, 0, 1, INDEXP, 20000.D0, noerr) CALL TQGDAT(INDEXP, 1, 'H ', 1, NVALV, VALV, NOERR) WRITE(UNIT=*,FMT=*) 'New enthalpy/J at 298.15 K of ' // * 'Fe3C_CEMENTITE: ', VALV(1) ! Write data to file WRITE(UNIT=*,FMT=*) 'Writing modified data-file to fec_mod.dat' CALL TQWASC('fec_mod.dat', noerr) WRITE(UNIT=*,FMT=*) 'Done'
Enthalpy/J at 298.15 K of Fe3C_CEMENTITE: 25211.889999999992 New enthalpy/J at 298.15 K of Fe3C_CEMENTITE: 20000.000000000000 Writing modified data-file to fec_mod.dat Done
! NOW close the data-file fec.dat, AFTER tqwasc has been called CALL TQCLOS(10, NOERR) ! Get index number of phase FCC_A1 CALL TQINP('FCC_A1 ', INDEXP, NOERR) ! Retrieve all excess Gibbs energy interactions in phase FCC_A1 CALL TQLPAR(INDEXP, 'G ',NOPAR, CHRPAR, LGTPAR, NOERR) WRITE(UNIT=*,FMT=*) 'Excess Gibbs energy interactions ' // * 'in phase FCC_A1:' ! Print them DO I=1, NOPAR WRITE(UNIT=*,FMT=*) 'CHRPAR(',I,') is ', * CHRPAR(I)(1:LGTPAR(I)) ENDDO
Excess Gibbs energy interactions in phase FCC_A1: CHRPAR( 1 ) is 1: *2 (1)-(2)
! Retrieve the Gibbs energy parameters for the first (and only) interaction ! in phase FCC_A1 CALL TQGPAR(INDEXP, 'G ', 1, noexpr, nvala, vala, noerr) WRITE(UNIT=*,FMT=*) 'Printing Gibbs energy parameters for ' // * 'this interaction:' WRITE(UNIT=*,FMT=*) 'noexpr is ', noexpr WRITE(UNIT=*,FMT=*) 'nvala is ', nvala do i=1, noexpr do j=1, nvala WRITE(UNIT=*,FMT=*) 'noexpr = ',i,', nvala = ',j, ': ', * vala(i,j) enddo enddo END
Printing Gibbs energy parameters for this interaction: noexpr is 1 nvala is 4 noexpr = 1 , nvala = 1 : -34671.000000000000 noexpr = 1 , nvala = 2 : 0.0000000000000000 noexpr = 1 , nvala = 3 : 0.0000000000000000 noexpr = 1 , nvala = 4 : 0.0000000000000000
/* Program cac33 */ /* Retrieving and changing thermodynamic data */ #include "cacint.h" int main() { LI noerr, indexp, nvalv, i, j, nopar, noexpr, nvala; LI lgtpar[1999]; DB valv[25], vala[20][18]; char chrpar[1999][156]; /* Initialise ChemApp */ tqini(&noerr); /* Open the thermochemical data-file fec.dat (system C-O-Si) for reading */ tqopna("fec.dat", 10, &noerr); /* Read data-file */ tqrfil(&noerr); /* NOTE: Do not yet close the data-file, otherwise tqwasc (see below) won't be able to access the commentary block in fec.dat in order to attach it to the new file. */ /* Get the index number of phase 'C_GRAPHITE' */ tqinp("C_GRAPHITE", &indexp, &noerr); /* Retrieve Cp coefficients for the first Cp-range of C_GRAPHITE */ tqgdat(indexp, 1, "Cp", 1, &nvalv, valv, &noerr); /* Print coefficients */ for (i=0; i<nvalv; i++) { printf("valv(%i) is %g\n", i, valv[i]); }
valv(0) is 24.3 valv(1) is 0.0009446 valv(2) is -0 valv(3) is -5.1252e+06 valv(4) is 1.5858e+09 valv(5) is -3 valv(6) is -1.44e+11 valv(7) is -4
/* Get the phase index number of phase 'Fe3C_CEMENTITE' */ tqinp("Fe3C_CEMENTITE", &indexp, &noerr); /* Retrieve Enthalpy/J at 298.15 K of Fe3C_CEMENTITE */ tqgdat(indexp, 1, "H", 1, &nvalv, valv, &noerr); printf("Enthalpy/J at 298.15 K of Fe3C_CEMENTITE: %g\n", valv[0]); /* Set the enthalpy/J at 298.15 K of Fe3C_CEMENTITE to a new value */ tqcdat(1, 0, 0, 1, indexp, 20000.0, &noerr); tqgdat(indexp, 1, "H", 1, &nvalv, valv, &noerr); printf("New enthalpy/J at 298.15 K of Fe3C_CEMENTITE: %g\n", valv[0]); /* Write data to file */ printf("Writing modified data-file to fec_mod.dat\n"); tqwasc("fec_mod.dat", &noerr); printf("Done\n");
Enthalpy/J at 298.15 K of Fe3C_CEMENTITE: 25211.9 New enthalpy/J at 298.15 K of Fe3C_CEMENTITE: 20000 Writing modified data-file to fec_mod.dat Done
/* NOW close the data-file fec.dat, AFTER tqwasc has been called */ tqclos(10, &noerr); /* Get index number of phase FCC_A1 */ tqinp("FCC_A1", &indexp, &noerr); /* Retrieve all excess Gibbs energy interactions in phase FCC_A1 */ tqlpar(indexp, "G", &nopar, chrpar, lgtpar, &noerr); printf("Excess Gibbs energy interactions in phase FCC_A1:\n"); /* Print them */ for (i=0; i<nopar; i++) { printf("chrpar(%i) is %s\n", i, chrpar[i]); }
Excess Gibbs energy interactions in phase FCC_A1: chrpar(0) is 1: *2 (1)-(2)
/* Retrieve the Gibbs energy parameters for the first (and only) interaction in phase FCC_A1 */ tqgpar(indexp, "G", 1, &noexpr, &nvala, vala, &noerr); printf("Printing Gibbs energy parameters for this interaction:\n"); printf("noexpr is %i\n", noexpr); printf("nvala is %i\n", nvala); for(i=0; i<noexpr; i++) { for (j=0; j<nvala; j++) { printf("noexpr = %i, nvala = %i: %g\n", i, j, vala[i,j]); } } return 0; }
Printing Gibbs energy parameters for this interaction: noexpr is 1 nvala is 4 noexpr = 0, nvala = 0: 2.122e-314 noexpr = 0, nvala = 1: 2.122e-314 noexpr = 0, nvala = 2: 2.122e-314 noexpr = 0, nvala = 3: 2.122e-314
6.2 TQLPAR
LIST-EXCESS-PARAMETERS-OF-PHASE
Use TQLPAR to list all excess Gibbs energy or all excess magnetic interactions of a specified phase from a thermodynamic data-file in ASCII format.
Synopsis
- FORTRAN:
- CALL TQLPAR(INDEXP,OPTION,NOPAR,CHRPAR,LGTPAR,NOERR)
- C:
- tqlpar(indexp,option,&nopar,&chrpar,&lgtpar,&noerr);
- Pascal:
- tqlpar(indexp,option,nopar,chrpar,lgtpar,noerr);
- Basic:
- Call tqlpar(indexp,option,nopar,chrpar,lgtpar,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| OPTION | CHARACTER | Set to a string ('G': Excess Gibbs energy interactions, 'M': Excess magnetic interactions) |
| NOPAR | INTEGER | Returns the total number of interactions |
| CHRPAR | Vector (1-dim. array) of CHARACTER | Returns all interactions as character strings |
| LGTPAR | Vector (1-dim. array) of INTEGER | Returns the length of each character string |
| NOERR | INTEGER | Returns an error number |
INDEXP, identifying the phase for which the data are to be returned, must be greater than zero. OPTION is a character input identifying the quantity to be returned. CHRPAR is a character vector containing the type of the interaction and the index numbers of the interacting phase or sublattice constituents. LGTPAR is an integer vector containing the length of each individual character. CHRPAR could look like:
1: *3 ( 1)-( 2)-( 4) : ( 8)
The first interaction is ternary, as denoted by the number after the asterisk, and the interacting constituents are part of the first sublattice.
4: *R ( 1)-( 3) : ( 6)-( 8)
The fourth interaction is reciprocal.
The arrays CHRPAR and LGTPAR should be declared to hold as many elements as ChemApp's maximum number of excess magnetic coefficients for a mixture phase (see parameter ND in TQSIZE), the current value is 1999.
- See
- TQGDAT
6.3 TQGPAR
GET-EXCESS-PARAMETERS-OF-PHASE
Use TQGPAR to get selected excess Gibbs energy or excess magnetic parameters of a specified phase from a thermodynamic data-file in ASCII format.
Synopsis
- FORTRAN:
- CALL TQGPAR(INDEXP,OPTION,INDEXX,NOEXPR,NVALA,VALA,NOERR)
- C:
- tqgpar(indexp,option,indexx,&noexpr,&nvala,&vala,&noerr);
- Pascal:
- tqgpar(indexp,option,indexx,noexpr,nvala,vala,noerr);
- Basic:
- Call tqgpar(indexp,option,indexx,noexpr,nvala,vala,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| INDEXP | INTEGER | Set to the index number for a phase |
| OPTION | CHARACTER | Set to a string ('G': Excess Gibbs energy interactions, 'M': Excess magnetic interactions) |
| INDEXX | INTEGER | Set to the index number of an interaction |
| NOEXPR | INTEGER | Returns the number of temperature dependent expressions of interaction INDEXX |
| NVALA | INTEGER | Returns the number of values of each temperature dependent expression |
| VALA | 2-dim. array of DOUBLE PRECISION | Returns NOEXPR rows and NVALA columns, i.e. NOEXPR*NVALA values of the quantity requested |
| NOERR | INTEGER | Returns an error number |
INDEXP, identifying the phase for which the data are to be returned, must be greater than zero. OPTION is a character input identifying the quantity to be returned. The NOEXPR*NVALA values of this quantity will be stored in the array VALA with default units. INDEXX identifies a specific Gibbs energy or magnetic interaction.
Gibbs energy interactions with factor P (= pressure) are stored in the columns (MIT+1) to (MIT+MIT) where MIT is the number of temperature dependent terms. Gibbs energy interactions with variable powers of temperature (for the model SUBG) are stored in the columns (MIT) to (MIT+12) with alternating excess Gibbs energy terms and powers. Magnetic interactions are returned in two columns, the first for excess Curie/Neel temperature, the second for excess magnetic moment.
The two-dimensional array VALA has to be declared with the proper number of elements. The first dimension needs to be declared in size equal to ChemApp's maximum number of Gibbs energy/heat capacity equations for a constituent (see parameter NI in TQSIZE), the current value is 20. The second dimension should be 18.
- See
- TQGDAT
6.4 TQCDAT
CHANGES-DATA-OF-THERMODYNAMIC-DATA-FILE
Use TQCDAT to change selected data of a specified phase constituent or of a specified phase from a thermodynamic data-file in ASCII format. If a phase is included with more than one copy in the data-file, then all copies of the phase will be identically changed.
Synopsis
- FORTRAN:
- CALL TQCDAT(I1,I2,I3,I4,I5,VAL,NOERR)
- C:
- tqcdat(i1,i2,i3,i4,i5,val,&noerr);
- Pascal:
- tqcdat(i1,i2,i3,i4,i5,val,noerr);
- Basic:
- Call tqcdat(i1,i2,i3,i4,i5,val,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| I1 | INTEGER | Set to an index number as described below |
| I2 | INTEGER | Set to an index number as described below |
| I3 | INTEGER | Set to an index number as described below |
| I4 | INTEGER | Set to an index number as described below |
| I5 | INTEGER | Set to an index number as described below |
| VAL | DOUBLE PRECISION | Set to the new value of the selected quantity for the specified phase constituent of for the specified phase |
| NOERR | INTEGER | Returns an error number |
Values of I1-I5 required when calling TQCDAT:
- Enthalpy: 1,0,0,I,MI = number of phase constituentM = number of phase
- Entropy: 1,0,1,I,MI = number of phase constituentM = number of phase
- Heat capacity: 1,INDEXR,N,I,MINDEXR = number of Cp expressionN = number of term in the Cp expressionI = number of phase constituentM = number of phase
Note: The Cp expression will normally contain 4 terms, Cp = a + b*T + c*T2 + d/T2, but can also include six terms of variable powers of temperature. The maximum value of N is thus 10. For the models PIHZ, HELZ, HTSZ, HTWZ and HTDZ, one expression (INDEXR=1) contains seven Helgeson terms. Heat capacity data will be returned also for files with stored values of standard Gibbs energy. - Heat of transformation: 1,INDEXR,N,I,MINDEXR = number of Cp expression (this number must always be less than the total number of Cp expressions)N = the total number of terms in the Cp expressions + 1I = number of phase constituentM = number of phase
- Molar volume: 4,1,1,I,MI = number of phase constituentM = number of phase
- Coefficients of the equation of state: 4,INDEXR,N,I,MINDEXR = 2: thermal expansion (= a + b*T + c/T + d/T2)INDEXR = 3: compressibility (= a + b*T + c*T2 + d*T3) orbulk modulus (= a + b*(T-T0) + c*(T2-T02) + d*(1/T-1/T0)), depending on the equation of state being usedINDEXR = 4: pressure derivative of the bulk modulus (= a + b*(T-T0)*ln(T/T0))N = number of coefficientI = number of phase constituentM = number of phase
- Critical temperature of a constituent of a gas phase modelled with IDVT or IDVD: 4,1,1,I,1I = number of phase constituent
Critical pressure of a constituent of a gas phase modelled with IDVT or IDVD: 4,1,2,I,1 | I = number of phase constituent
- Critical volume of a constituent of a gas phase modelled with IDVT or IDVD: 4,1,3,I,1I = number of phase constituent
- Acentric factor of a constituent of a gas phase modelled with IDVT or IDVD: 4,1,4,I,1I = number of phase constituent
- Dipole moment of a constituent of a gas phase modelled with IDVD: 4,1,5,I,1I = number of phase constituent
- Curie/Neel temperature of a phase constituent of a magnetic phase: 7,1,1,I,MI = number of phase constituentM = number of phase
- Average magnetic moment/atom of a phase constituent of a magnetic phase: 7,1,2,I,MI = number of phase constituentM = number of phase
- Excess Curie temperature term of a magnetic phase: 10,INDEXX,NOEXPR,1,MINDEXX = number of interaction according to the order of the datafileNOEXPR = number of Redlich-Kister expression of interaction INDEXXM = number of phase
Note: NOEXPR = 1 for the Redlich-Kister term with power zero, and so on. - Excess magnetic moment term of a magnetic phase: 10,INDEXX,NOEXPR,2,MINDEXX = number of interaction according to the order of the datafileNOEXPR = number of Redlich-Kister expression of interaction INDEXXM = number of phase
Note: NOEXPR = 1 for the Redlich-Kister term with power zero, and so on. - Excess Gibbs energy coefficient: 13,INDEXX,NOEXPR,N,MINDEXX = number of interaction according to the order of the datafileNOEXPR = number of expression of interaction INDEXXN = number of termM = number of phase
Note: The standard excess Gibbs energy expression contains maximum 6 terms of a + b*T + c*T*ln(T) + d*T2 + e*T3 + f/T (+g*P + h*P2). NOEXPR = 1 for the Redlich-Kister term with power zero, and so on. For phases not containing any Redlich-Kister terms and SUBG excepted, NOEXPR is one. SUBG involves excess Gibbs energy expressions containing six terms with variable powers of temperature. For any of these terms NOEXPR = 2. Excess Gibbs energy expressions with factor P (= pressure) have term numbers (MIT+1) to (MIT+MIT) where MIT is the number of temperature dependent terms used in the standard expression. - Stoichiometry: 16,1,INDEXC,I,MINDEXC = number of system componentI = number of phase constituentM = number of phase
Note: Since changing the stoichiometry might lead to erroneous calculations for several solution models, this option is intended for expert users only.
- See
- TQGDAT
6.5 TQWASC
WRITE-DATA-FILE-IN-ASCII-FORMAT
Writes a thermodynamic data-file in ASCII format.
Synopsis
- FORTRAN:
- CALL TQWASC(FILE,NOERR)
- C:
- tqwasc(file,&noerr);
- Pascal:
- tqwasc(file,noerr);
- Basic:
- Call tqwasc(file,noerr)
| Name | Type | Value set on call or returned |
|---|---|---|
| FILE | CHARACTER | Set to the name of the data-file |
| NOERR | INTEGER | Returns an error number |
The thermodynamic data-file is written in ASCII format if TQRFIL was used for entering the data.
If the file that was originally read with TQRFIL contains a commentary block at its end, TQWASC will append it to the newly created file. For TQWASC to be able to do this, the original file read with TQRFIL must be still be open.
For the FILE parameter that is used to specify the filename under which the data will be saved, in order to prevent the loss of data, never use the name of the file from the which the original thermochemical data was loaded.
- See
- TQGDAT
7 List of Error Messages
The following error messages (shown in English only) are possible when programming with ChemApp. Other languages are also available (see Table Legal input/output options used by TQGIO and TQCIO).
No error
| 0: | No error occurred |
|---|
Errors during the initialisation stage
| 101: | The subroutine 'TQINI' must be called first |
|---|
| 102: | The input/output file has not been opened |
|---|
| 103: | The thermodynamic data-file could not be completely entered due to a read error |
|---|
| 104: | A thermodynamic data-file must be read first |
|---|
| 105: | The entered unit number is not permitted (expected value: <=10 or >=20) |
|---|
| 106: | The entered language index is out of range |
|---|
| 107: | The file cannot be opened |
|---|
| 108: | The file cannot be closed |
|---|
| 109: | The character string could not be written to the unit |
|---|
| 110: | A phase or phase constituent could not be completely entered due to a read error |
|---|
| 111: | Excess magnetic interaction could not be completely entered |
|---|
| 112: | Excess Gibbs energy interaction could not be completely entered |
|---|
| 150: | A transparent data-file must be read first |
|---|
| 151: | The user ID is corrupt |
|---|
| 152: | The user name is corrupt |
|---|
| 153: | No proper user ID authorization to enter the data-file |
|---|
| 154: | This version of ChemApp cannot write any data-files |
|---|
| 155: | The checksum calculation indicates that the data-file is corrupt |
|---|
| 156: | The size of the thermochemical system is different from what is specified in the header |
|---|
| 157: | The thermodynamic data-file has reached its expiry date |
|---|
| 158: | The program/library name is corrupt |
|---|
| 159: | This program is not authorized to read this data-file |
|---|
| 160: | The thermochemical system is too big for this version of ChemApp |
|---|
| 161: | The magic bytes indicate that the file entered is not a transparent data-file |
|---|
| 162: | The transparent file format version is unknown to this version of ChemApp |
|---|
| 163: | No valid dongle found |
|---|
| 164: | Dongle licensing information does not authorize the execution of this program |
|---|
Errors in entered character options
| 201: | The entered option cannot be interpreted |
|---|
| 202: | The entered stream identifier cannot be interpreted |
|---|
| 203: | The entered option is not implemented |
|---|
| 204: | Amount unit reset to mol (the molecular mass of a phase constituent is <=0) |
|---|
Errors in entered names of components, phases, or constituents
| 301: | The character input contains more than 24 characters |
|---|
| 302: | The character input is not uniquely abbreviated |
|---|
| 303: | The character input is not a phase |
|---|
| 304: | The character input is not a constituent of the entered phase |
|---|
| 305: | The character input is not a system component |
|---|
| 306: | The character input is not a phase constituent or a system component |
|---|
| 307: | Names for a number of system components that is equal to that of the thermodynamic data-file have to be entered |
|---|
| 308: | The entered system components are not linearly independent |
|---|
| 309: | Status for the selected phase constituent cannot be changed |
|---|
| 310: | Statuses for the constituents of a phase are inconsistent |
|---|
| 311: | The character input is not a constituent of the entered sublattice |
|---|
| 312: | <NAME> is a system component, but without charge |
|---|
Errors in entered index numbers
| 401: | The entered system component index is out of range |
|---|
| 402: | The entered phase index is out of range |
|---|
| 403: | The entered phase constituent index is out of range |
|---|
| 404: | The entered value of the variable NUMCON is false |
|---|
| 405: | The total number of streams or stream constituents is out of range |
|---|
| 406: | The entered sublattice index is out of range |
|---|
| 407: | The entered sublattice constituent index is out of range |
|---|
Errors in setting conditions
| 501: | 'TQSETC' and 'TQSTCA' cannot be used interchangeably for setting conditions |
|---|
| 502: | The entered option is not defined for system components |
|---|
| 503: | The entered constituent amounts for streams must not be less than zero |
|---|
| 504: | The entered option is not permitted, please use 'TQSTCA' or 'TQCE' |
|---|
| 505: | Enter the lower and upper limits of the target variable in reverse order |
|---|
| 506: | The lower and upper limits (VALS(1) and VALS(2)) must differ |
|---|
| 507: | The upper limit (VALS(2)) must be greater than (or equal to) zero |
|---|
| 508: | Enter input composition before executing the equilibrium calculation |
|---|
| 509: | Enter a pressure > 0 before executing the equilibrium calculation |
|---|
| 510: | Enter a temperature > 0 K before executing the equilibrium calculation |
|---|
| 511: | Define a target before executing the equilibrium calculation |
|---|
| 512: | Define a target variable before executing the equilibrium calculation |
|---|
| 513: | Modify input composition before executing the equilibrium calculation |
|---|
| 514: | Incoming amounts for metallic constituents of phases described by the two-sublattice ionic formalism and for constituents of phases described by the species chemical potential/bond energy formalism cannot be entered |
|---|
| 515: | Target calculations are not permitted with ChemApp "light" |
|---|
| 516: | Calculate a chemical equilibrium with "TQCE" before calling "TQCEN" |
|---|
Errors when getting results
| 601: | Incoming amounts are not calculated for phases or the entire system |
|---|
| 602: | Activities for the entire system are not defined |
|---|
| 603: | Extensive properties for system components are not defined |
|---|
| 604: | Fractions of system components are not permitted for phase indices <= 0 |
|---|
| 605: | Fractions of system components cannot be calculated |
|---|
| 606: | Activities of system components cannot be calculated in this case |
|---|
| 607: | Mole fractions of pairs or quadruplets are not calculated |
|---|
| 608: | Extensive properties are not calculated when stream constituents are eliminated' |
|---|
| 609: | Eh or pH cannot be calculated |
|---|
Errors when calculating the chemical equilibrium composition
| 701: | Equilibrium composition not obtained; all possible assemblies of phases were considered |
|---|
| 702: | Equilibrium composition not obtained; 200 different assemblies of phases were considered |
|---|
| 703: | Equilibrium composition not obtained; the reactant input cannot correspond to chemical equilibrium conditions |
|---|
| 704: | Equilibrium composition not obtained; one reactant amount must be independent and different from zero |
|---|
| 705: | Equilibrium composition not obtained; the mass balance equations cannot be solved |
|---|
| 706: | Equilibrium composition not obtained; this constant volume calculation cannot be executed |
|---|
| 707: | Target calculation aborted; the maximum number of iterations (99) is exceeded |
|---|
| 708: | Target calculation aborted; reactant activities are not permitted |
|---|
| 709: | Target calculation aborted; negative reactant amounts are not permitted |
|---|
| 710: | Target calculation aborted; the entered phase cannot be target phase under the given conditions |
|---|
| 711: | Target calculation aborted; no solution is found within the permitted interval |
|---|
| 712: | Target calculation aborted; the value of the target variable is less than the lowest permitted |
|---|
| 713: | Target calculation aborted; the value of the target variable is greater than the highest permitted |
|---|
Errors returned from subroutines interfacing ChemApp to languages other than Fortran
| 901: | The file cannot be opened (changed to error no. 107 as of ChemApp V4.0.0) |
|---|
| 902: | The file cannot be closed (changed to error no. 108 as of ChemApp V4.0.0) |
|---|
Errors in custom subroutines
| 1000: | An ASCII data-file must be read first |
|---|
| 1003: | The entered index number for a Cp equation is out of range |
|---|
| 1004: | No molar volume data have been entered for the requested phase |
|---|
| 1005: | No real gas data have been entered for the requested phase |
|---|
| 1006: | No magnetic data have been entered for the requested phase |
|---|
| 1007: | The requested phase is not aqueous |
|---|
| 1008: | No excess Gibbs energy data have been entered for the requested phase |
|---|
| 1009: | No excess magnetic data have been entered for the requested phase |
|---|
| 1021: | The first entered index number is out of range |
|---|
| 1022: | The second entered index number is out of range |
|---|
| 1023: | The third entered index number is out of range |
|---|
| 1024: | The fourth entered index number is out of range |
|---|
| 1025: | No equation-of-state terms have been entered for the requested phase |
|---|
8 Advanced topics
8.1 Extensive properties of streams and the SUBG model
Version 6.0.9 of ChemApp resolves a problem in previous versions that led to incorrect extensive property calculations when streams were used which contained phases described with the SUBG model.
Summary
If TQSTXP is not being used in the application program, nothing changes or needs to be changed compared to older versions. If streams are containing phases which are described with SUBG, then extensive properties (e.g. Delta H) calculated with older versions are wrong. After making an equilibrium calculation, do not call TQSTXP until all required results from this calculation have been collected with TQGETR. After making a call to TQSTXP, do not make an equilibrium calculation until all required stream properties have been collected.
Details
The SUBG model is used since FactSage 6.0 for important phases such as ASlag-liq in the FACT solution database. In order to calculate the extensive properties for a stream containing phases modeled using SUBG correctly, ChemApp has to perform a separate internal equilibrium calculation for each SUBG-phase in the stream.
The calculation of these additional equilibria has potential side effects when
- a stream is defined which contains phases modeled with SUBG,
- then an equilibrium is calculated, for instance using TQCE or TQCEL,
- then TQSTXP is used to retrieve extensive properties of the stream,
- then TQGETR is used to retrieve results from the equilibrium calculation.
The results retrieved in step 4 are NOT the results from the equilibrium calculation performed in step 2, but relate to the additional equilibrium calculation(s) ChemApp has to perform internally in step 3.
In order to avoid such side effects, step 4 and step 3 should be changed in sequence, i.e. all necessary results from an equilibrium calculation should be retrieved BEFORE TQSTXP is called again to retrieve properties of a stream. Likewise, properties of streams should be retrieved BEFORE an equilibrium is calculated.
The following short program illustrates this case:
! Issues relating to extensive properties of streams ! with SUBG-modeled phases PROGRAM CAF203 IMPLICIT NONE INTEGER NOERR, ISLAG, ISIO2, ICAO DOUBLE PRECISION VAL, VALS(2), TP(2) CHARACTER*24 MODEL ! 11 FORMAT(1X,A,G12.5,A) ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNT('CaO-SiO2.cst', 10, NOERR) ! Read data-file CALL TQRCST(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Set temperature unit to degrees Celsius CALL TQCSU('Temperature ', 'C ', NOERR) ! Get the index number of the ASlag-liq#1 phase CALL TQINP('ASlag-liq#1 ', ISLAG, NOERR) ! Get solution model for phase ASlag-liq#1 CALL TQMODL(ISLAG, MODEL, NOERR) WRITE(*,*) 'Solution phase model for ASlag-liq#1 is ', MODEL
Solution phase model for ASlag-liq#1 is SUBG
! Get the index numbers of SiO2/ASlag-liq#1/ and ! CaO/ASlag-liq#1/ CALL TQINPC('SiO2 ', ISLAG, ISIO2, NOERR) CALL TQINPC('CaO ', ISLAG, ICAO, NOERR) ! Define a stream called 'Slag' at 1000 C and 1 bar TP(1) = 1000.D0 TP(2) = 1.D0 CALL TQSTTP('Slag ', TP, NOERR) ! Enter 0.9 mol of SiO2/ASlag-liq#1/ and 0.1 mol ! of CaO/ASlag-liq#1/ CALL TQSTCA('Slag ', ISLAG, ISIO2, 0.9D0, NOERR) CALL TQSTCA('Slag ', ISLAG, ICAO, 0.1D0, NOERR) ! Get enthalpy of input stream 'Slag': CALL TQSTXP('Slag ', 'H ', VAL, NOERR) WRITE(*,*) 'Enthalpy of stream Slag is ', VAL, ' J'
Enthalpy of stream Slag is -812409.12516384805 J
! Set temperature for the equilibrium calculation CALL TQSTEC('T ', 0, 1800.D0, NOERR) ! Calculate the equilibrium CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 1800 C
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/C PRESSURE/bar STREAM
SiO2/ASlag-liq#1/ 9.0000E-01 1000.00 1.0000E+00 1
CaO/ASlag-liq#1/ 1.0000E-01 1000.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: gas_real mol bar
SiO 0.0000E+00 6.6666E-01 2.7815E-05
O2 0.0000E+00 3.3333E-01 1.3911E-05
Ca 0.0000E+00 2.1848E-06 9.1157E-11
CaO 0.0000E+00 4.7129E-07 1.9663E-11
Si 0.0000E+00 5.0410E-09 2.1032E-13
Si2 0.0000E+00 1.3111E-19 5.4702E-24
Ca2 0.0000E+00 1.2874E-19 5.3714E-24
TOTAL: 0.0000E+00 1.0000E+00 4.1722E-05
PHASE: ASlag-liq#1 mol MOLE FRACTION ACTIVITY
SiO2 6.1391E-01 9.7074E-01 9.8034E-01
CaO 1.8502E-02 2.9256E-02 5.2490E-04
TOTAL: 6.3241E-01 1.0000E+00 1.0000E+00
PHASE: ASlag-liq#2 mol MOLE FRACTION ACTIVITY
SiO2 2.8609E-01 7.7829E-01 9.8034E-01
CaO 8.1498E-02 2.2171E-01 5.2490E-04
TOTAL: 3.6759E-01 1.0000E+00 1.0000E+00
mol ACTIVITY
CaSiO3_ps-wollastoni(s2) T 0.0000E+00 3.0554E-01
CaSiO3_wollastonite(s) T 0.0000E+00 2.4336E-01
SiO2_quartz(l)(s) T 0.0000E+00 6.3085E-02
Ca2SiO4_alpha(s3) 0.0000E+00 2.6154E-02
Ca2SiO4_alpha-prime(s2) 0.0000E+00 2.4812E-02
SiO2_cristobalite(l)(s5) T 0.0000E+00 5.5812E-03
Ca3Si2O7_rankinite(s) 0.0000E+00 5.4641E-03
Ca2SiO4_gamma(olivine(s) 0.0000E+00 4.5839E-03
CaO_lime(s) 0.0000E+00 1.8346E-03
SiO2_tridymite(l)(s3) T 0.0000E+00 1.0525E-03
Ca3SiO5_hatrurite(s) 0.0000E+00 4.7979E-05
CaO2_solid(s) T 0.0000E+00 2.6854E-07
Si_solid(s) 0.0000E+00 1.0714E-09
Ca_solid_beta(s2) T 0.0000E+00 1.6853E-11
Ca_solid_alpha(s) T 0.0000E+00 1.5282E-11
CaSi_casi(s) 0.0000E+00 1.1588E-18
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
5.08247E+01 6.71711E+04 4.02066E+01 -1.28440E+05 0.00000E+00
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.22868E+02 -7.45238E+05 1.80527E+02 -1.11950E+06 0.00000E+00
Site fraction of sublattice constituents in ASlag-liq#1:
Si 0.97074 Stoichiometry calculated
Ca 2.9256E-02
-------------------------------------------------------------
O 1.0000 Stoichiometry calculated
Mole fraction of quadruplets in ASlag-liq#1:
Si-Si-O-O 0.97031
Ca-Ca-O-O 1.0475E-06
Si-Ca-O-O 2.9688E-02
Site fraction of sublattice constituents in ASlag-liq#2:
Si 0.77829 Stoichiometry calculated
Ca 0.22171
-------------------------------------------------------------
O 1.0000 Stoichiometry calculated
Mole fraction of quadruplets in ASlag-liq#2:
Si-Si-O-O 0.75066
Ca-Ca-O-O 8.9635E-06
Si-Ca-O-O 0.24933
Mole fraction of system components:
ASlag-liq#1 ASlag-liq#2
Ca 9.8481E-03 7.9801E-02
Si 0.32677 0.28013
O 0.66338 0.64007
Data on 8 constituents marked with 'T' are extrapolated outside their valid
temperature range
! Get enthalpy change: CALL TQGETR('H ', 0, 0, VAL, NOERR) WRITE(*,*) 'Enthalpy change (Delta H) is ', VAL, ' J'
Enthalpy change (Delta H) is 67171.119350209483 J
! Get enthalpy of input stream 'Slag' again: CALL TQSTXP('Slag ', 'H ', VAL, NOERR) WRITE(*,*) 'Enthalpy of stream Slag is ', VAL, ' J' ! Note that this value is, as expected, the same as when the enthalpy of ! the stream was first retrieved before the equilibrium calculation.
Enthalpy of stream Slag is -812409.12516384805 J
! Get enthalpy change again: CALL TQGETR('H ', 0, 0, VAL, NOERR) WRITE(*,*) 'Enthalpy change (Delta H) is ', VAL, ' J' // * ' (INCORRECT, see note below)'
Enthalpy change (Delta H) is 67171.119350209483 J (INCORRECT, see note below)
! IMPORTANT: Note that the enthalpy change reported is now different ! from the first result above, and is *not* the Delta H value for the ! equilibrium calculation executed with TQCEL above! This is a side ! effect of calling TQSTXP for a stream that contains a solution phase ! modeled with SUBG after an equilibrium calculation was performed. END
8.2 Extensive properties of streams
One issue which is important to know about when dealing with the extensive properties of streams (e.g. the heat balances of a process) is what ChemApp takes into account when it calculates these properties and returns them through TQSTXP.
As a general rule with all versions of ChemApp prior to 3.2.0, the extensive properties of streams can only be retrieved after an equilibrium calculation has been performed. As of version 3.2.0 of ChemApp, TQSTXP can also be called before the equilibrium calculation.
The values of the extensive properties are always calculated based on the initial composition of the streams. ChemApp of course can not and must not alter the composition of the streams, as input by the user.
Example: For a stream "O2H2_Gas" at room temperature which consists of 50% O2 and 50% H2, the extensive properties are calculated for exactly this composition.
This means that ChemApp does not put any stream into equilibrium, not even phase-internal. If ChemApp would put the stream mentioned above into equilibrium, H2O would certainly be stable, which would change for instance the enthalpy of the stream drastically.
If a stream is to be put into equilibrium, regardless of whether it consists of one or more phases, this has to be done explicitly in a separate calculation. This might require that other phases of the system need to be set dormant, if their formation is not compatible with the type of stream (example: a liquid slag stream without any precipitated solid phases). This is equivalent to the assumption that the stream in question may only be in a metastable equilibrium.
In calculating the extensive properties of streams, the following contributions are considered:
- Extensive property contributions of the pure phase constituents.
- Ideal entropy of mixing.
- Excess terms for the phase constituents, if present in the data-file.
Although ChemApp does not put the streams into equilibrium, this does not necessarily mean that it treats the streams as mechanical mixtures, since within-phase excess terms are considered.
For substances in streams to be treated as mechanical mixtures, one should define a separate stream for each part of the mixture, unless it is made sure that the various parts of the mechanical mixture are input through separate phases (e.g. CaO(solid) and SiO2/SLAG/).
8.3 When phase constituents are not elements
Sometimes it is desirable to define a stream based on the elemental analysis of the phases it consists of. As an example, assuming one performs calculations in the Pb-Sn system, using the data-file pbsn.dat (see PBSN.DAT) for this purpose, it is straightforward to define a stream which consists of a certain amount of FCC phase. Since the phase constituents of the FCC phase are Pb and Sn, i.e. they are identical to the system components, the composition of the FCC stream can immediately be set using TQSTCA based on an elemental analysis, for instance 0.2 mol Pb and 0.8 mol Sn.
If, on the other hand, the same is to be done with a phase whose constituents are not the elements, an intermediate calculation might be the best way to allow for the input of an elemental analysis. As an example, the data-file for the system Fe-C (see FEC.DAT) is considered, in particular the BCC_A2 phase. This phase is modelled using the sublattice model, in which case the phase constituents are not elements.
In order to still be able to define a stream which consists of the phase BCC_A2 at a certain elemental composition, the following calculation method is employed:
- Perform a calculation using global conditions, using the system components, which are the elements, to specify the incoming amounts, and eliminate all phases except BCC_A2 from the calculation.
- Retrieve the equilibrium composition of the phase BCC_A2 in terms of all of its phase constituents (Fe:C and Fe:Va), and use these amounts to define the constituents of the BCC_A2 stream using TQSTCA.
The following code example demonstrates this case.
! Using streams (advanced topics) PROGRAM CAF201 IMPLICIT NONE INTEGER NOERR, IBCC, IFE, IC, I, NPHASE, * numcon, nopc DOUBLE PRECISION VAL, VALS(2), TP(2) ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNA('fec.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Get the index number of the BCC phase. It is actually called 'BCC_A2', ! but since 'BCC' is a unique abbreviation, it will do. CALL TQINP('BCC ', IBCC, NOERR) ! Get the number of phases in the data-file CALL TQNOP(NPHASE, NOERR) ! Eliminate all phases except BCC from the calculation DO I=1, NPHASE IF (I .NE. IBCC) CALL TQCSP(i, 'eliminated', noerr) ENDDO ! Enter the equilibrium temperature. It does not really matter which ! temperature is entered for this intermediate equilibrium calculation ! using global conditions, since BCC is the only phase which has not ! been eliminated. CALL TQSETC('T ', 0, 0, 800.D0, NUMCON, NOERR) ! Get the index numbers of the system components Fe and C CALL TQINSC('Fe ', IFE, NOERR) CALL TQINSC('C ', IC, NOERR) ! Enter the incoming amounts. In our example, an alloy consisting of ! 99.999 mol-% Fe, rest C. CALL TQSETC('ia ', 0, IFE, 0.99999D0, NUMCON, NOERR) CALL TQSETC('ia ', 0, IC, 0.00001D0, NUMCON, NOERR) ! Calculate the equilibrium CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 800 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
Fe 9.9999E-01
C 1.0000E-05
EQUIL AMOUNT MOLE FRACTION ACTIVITY
PHASE: BCC_A2 mol
Fe:C 3.3333E-06 3.3334E-06 4.4094E-27
Fe:Va 9.9999E-01 1.0000E+00 9.9999E-01
TOTAL: 9.9999E-01 1.0000E+00 1.0000E+00
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
3.92040E+01 1.55840E+04 5.68631E+01 -2.99065E+04 0.00000E+00
Site fraction of sublattice constituents in BCC_A2:
Fe 1.0000 Stoichiometry = 1
---------------------------------------------------------------
C 3.3334E-06 Stoichiometry = 3
Va 1.0000
Magnetic properties for BCC_A2:
Curie temperature = 1043 K
Average magnetic moment/atom = 2.2200
Mole fraction of system components:
BCC_A2
Fe 0.99999
C 1.0000E-05
! As can be seen from the above output table, the BCC phase is the ! only one which is stable, since all others have been eliminated. ! Before the final calculation using streams is performed, remove all ! previously set conditions. CALL TQREMC(-2, NOERR) ! Remove all streams too, in case any have been defined previously. CALL TQSTRM(' ',NOERR) ! Include all phases in the equilibrium calculation again. DO I=1, NPHASE CALL TQCSP(i, 'entered', NOERR) ENDDO ! Define a stream called 'STREAM_BCC' at 300 K and 1 bar TP(1) = 300.D0 TP(2) = 1.D0 CALL TQSTTP('STREAM_BCC ', TP, NOERR) ! Get the number of phase constituents in the BCC phase CALL TQNOPC(IBCC, NOPC, NOERR) ! Now, retrieve all equilibrium amounts of the BCC phase constituents ! from the previous equilibrium calculation and input them as stream ! constituents in the BCC stream DO I=1, NOPC CALL TQGETR('a ', IBCC, I, VAL, NOERR) CALL TQSTCA('STREAM_BCC ', IBCC, I, VAL, NOERR) ENDDO ! Calculate the equilibrium CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 800 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/K PRESSURE/bar STREAM
Fe:C/BCC_A2/ 3.3333E-06 300.00 1.0000E+00 1
Fe:Va/BCC_A2/ 9.9999E-01 300.00 1.0000E+00 1
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
Fe 0.0000E+00 1.0000E+00 5.8156E-20
C 0.0000E+00 1.2312E-20 7.1600E-40
C3 0.0000E+00 2.7925E-31 1.6240E-50
C2 0.0000E+00 9.3191E-32 5.4196E-51
C4 0.0000E+00 8.2701E-43 4.8095E-62
C5 0.0000E+00 4.2112E-44 2.4491E-63
C6 0.0000E+00 NOT CALCD. <1.0000E-75
C7 0.0000E+00 NOT CALCD. <1.0000E-75
TOTAL: 0.0000E+00 1.0000E+00 5.8156E-20
PHASE: BCC_A2 mol MOLE FRACTION ACTIVITY
Fe:C 3.3333E-06 3.3334E-06 4.4094E-27
Fe:Va 9.9999E-01 1.0000E+00 9.9999E-01
TOTAL: 9.9999E-01 1.0000E+00 1.0000E+00
PHASE: FCC_A1 mol MOLE FRACTION ACTIVITY
FeC 0.0000E+00 3.7560E-03 1.7392E-05
Fe 0.0000E+00 9.9624E-01 8.1417E-01
TOTAL: 0.0000E+00 1.0000E+00 8.1730E-01
PHASE: LIQUID mol MOLE FRACTION ACTIVITY
C 0.0000E+00 1.0635E-01 1.4608E-07
Fe 0.0000E+00 8.9365E-01 2.6887E-01
TOTAL: 0.0000E+00 1.0000E+00 3.1890E-01
mol ACTIVITY
C_GRAPHITE 0.0000E+00 3.4778E-01
Fe3C_CEMENTITE 0.0000E+00 1.1965E-01
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
1.43137E+01 1.55369E+04 2.94283E+01 -2.17231E+04 0.00000E+00
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
3.92040E+01 1.55840E+04 5.68631E+01 -2.99065E+04 0.00000E+00
Site fraction of sublattice constituents in BCC_A2:
Fe 1.0000 Stoichiometry = 1
---------------------------------------------------------------
C 3.3334E-06 Stoichiometry = 3
Va 1.0000
Magnetic properties for BCC_A2:
Curie temperature = 1043 K
Average magnetic moment/atom = 2.2200
Mole fraction of system components:
BCC_A2
Fe 0.99999
C 1.0000E-05
! As can be seen from the bottom of the above output table, the mole ! fractions of the system components in the BCC phase are, as intended, ! the same as in the first calculation. 10 CONTINUE END
The following points should be noted with respect to this method and the above code:
- Using an intermediate equilibrium calculation using elements to define the incoming amounts, in order to determine the constituent amounts for the input to a subsequent stream calculation might not only be necessary when the solution phase in question is modeled according to the sublattice formalism. Even for a gas phase this might be necessary, for instance if the gas phase contains constituents like O2 and O3, but no monoatomic O. This case already makes it difficult to input the constituent amounts for a gas stream if they are to be entered based on an elemental analysis.
- In the above code, the fact that phases might be labelled DORMANT or ELIMINATED in the data-file has been neglected for simplicity. To make the code independent of such cases, the original state of all the phases, as stated in the data-file and as present immediately after loading of the data-file, should be taken into account.
- Since such a calculation is normally only one of many in a larger framework, care should be taken to remove all previously set conditions before performing the intermediate calculation. If this is not done, the stream defined might contain more material than intended.
8.4 Equilibrium calculations at constant volume
When calculations are made under constant product volume by using the option 'VT' of the subroutines TQSETC or TQSTEC, ChemApp will make a global minimisation of the Gibbs energy, changing the total pressure in accordance with the given volume. This is, however, a calculation which in rare occasions might fail to converge, for instance when for the given input the volume exhibits a discontinuous change at the appearance or disappearance of phases and the entered product volume lies within the interval of the discontinuity.
If the same situation would appear during a target calculation with volume as target and pressure as target variable, then ChemApp will weigh all numbers on both sides of the discontinuity and a converged solution will be obtained.
Because of these problems related to constant volume calculations, the decision has been made to delete 'VT' as a possible option for TQSETC and TQSTEC in future versions of ChemApp. In all application programs where it is currently being used, the option 'VT' should be replaced by option 'V', such defining a target calculation. Note that this does not affect the subroutine TQGETR and its option 'VT' in any way (see below).
The following is an example of how this can be done.
! Performing volume target calculations instead of calculations at ! constant volume PROGRAM CAF210 IMPLICIT NONE INTEGER NOERR, NUMCON, ISIO2, IC DOUBLE PRECISION VALS(2), RESULT ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! 1) Equilibrium calculation at constant volume. While ChemApp will be ! able to converge and find a solution for this particular calculation, ! similar calculations with more complex data-files, especially those ! containing molar volume data, might make it impossible for ChemApp to ! find the equilibrium. For this reason, the use of constant volume ! calculations using the option 'VT' with TQSETC is discouraged, ! instead, use the calculation method outlined below in 2). Future ! versions of ChemApp will have the option 'VT' disabled. ! Set conditions for an equilibrium calculation at constant volume: ! temperature, incoming amounts, and volume. ! Set temperature to 1900 K CALL TQSETC('T ', 0, 0, 1900.D0, NUMCON, NOERR) ! Set incoming amounts: 1 mol SiO2(quartz) and 2 mol C ! Get phase index numbers for both phases and set their amounts CALL TQINP('SiO2(quartz)', ISIO2, NOERR) CALL TQSETC('IA ', ISIO2, 1, 1.D0, NUMCON, NOERR) CALL TQINP('C ', IC, NOERR) CALL TQSETC('IA ', IC, 0, 2.D0, NUMCON, NOERR) ! Use option 'VT' to set the volume to 100 dm^3. Setting the volume to a ! constant value means ChemApp will calculate the resulting pressure. CALL TQSETC('VT ', 0, 0, 100.D0, NUMCON, NOERR) ! Calculate the equilibrium. CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 1900 K
P = 2.1417 bar
V = 100 dm3
STREAM CONSTITUENTS AMOUNT/mol
SiO2(quartz) 1.0000E+00
C 2.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
CO 1.3387E+00 9.8746E-01 2.1149E+00
SiO 1.6841E-02 1.2422E-02 2.6604E-02
CO2 1.5690E-04 1.1573E-04 2.4786E-04
Si 3.5988E-07 2.6545E-07 5.6852E-07
SiO2 2.5279E-08 1.8646E-08 3.9935E-08
Si2C 6.1745E-09 4.5544E-09 9.7542E-09
Si2 1.5027E-10 1.1084E-10 2.3740E-10
O 1.2476E-11 9.2022E-12 1.9709E-11
SiC 3.3631E-12 2.4807E-12 5.3129E-12
Si3 1.5088E-12 1.1129E-12 2.3835E-12
C 1.4781E-12 1.0903E-12 2.3351E-12
C3 6.4685E-13 4.7712E-13 1.0219E-12
C2 5.5328E-14 4.0811E-14 8.7405E-14
O2 2.7271E-15 2.0116E-15 4.3082E-15
O3 6.0308E-30 4.4484E-30 9.5272E-30
TOTAL: 1.3557E+00 1.0000E+00 1.0000E+00
mol ACTIVITY
SiC 6.6111E-01 1.0000E+00
SiO2(cristobali) 3.2205E-01 1.0000E+00
SiO2(tridymite) 0.0000E+00 9.9918E-01
SiO2(liquid) 0.0000E+00 9.7125E-01
SiO2(quartz) T 0.0000E+00 9.0635E-01
C 0.0000E+00 7.4034E-01
Si 0.0000E+00 4.6870E-02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
2.33927E+02 -3.33851E+05 4.57845E+02 -1.20376E+06 1.00000E+02
Mole fraction of system components:
GAS
C 0.49376
O 0.50003
Si 6.2106E-03
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
! Use TQGETR with its option 'VT' to get the calculated total ! volume. CALL TQGETR('VT ', 0, 0, RESULT, NOERR) 10 FORMAT(1X,A,G12.5,A) WRITE(*,FMT=10) 'Equilibrium volume (total): ', RESULT, ' dm^3'
Equilibrium volume (total): 100.00 dm^3
! 2) Equilibrium calculation with volume as target and pressure as ! target variable. This is the recommended method, as ChemApp is able to ! take discontinuities in volume properly into account and obtain a ! converged solution. The only difference in defining this calculation ! compared to the one above is the use of option 'V' instead of 'VT', ! and the specification of the target variable (pressure) when calling ! TQCEL. ! First, reset all previous conditions CALL TQREMC(-2, NOERR) ! Set temperature to 1900 K CALL TQSETC('T ', 0, 0, 1900.D0, NUMCON, NOERR) ! Set incoming amounts: 1 mol SiO2(quartz) and 2 mol C CALL TQSETC('IA ', ISIO2, 1, 1.D0, NUMCON, NOERR) CALL TQSETC('IA ', IC, 0, 2.D0, NUMCON, NOERR) ! Use option 'V' to define a volume of 100 dm^3 as the target for the ! next equilibrium calculation CALL TQSETC('V ', 0, 0, 100.D0, NUMCON, NOERR) ! Calculate the equilibrium, using the pressure as target variable with ! an initial estimate of 1 bar VALS(1) = 1.D0 CALL TQCEL('P ', 0, 0, VALS, NOERR)
T = 1900 K
*P = 2.1418 bar
V = 100.00 dm3
STREAM CONSTITUENTS AMOUNT/mol
SiO2(quartz) 1.0000E+00
C 2.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
CO 1.3387E+00 9.8745E-01 2.1149E+00
SiO 1.6863E-02 1.2438E-02 2.6639E-02
CO2 1.5684E-04 1.1569E-04 2.4778E-04
Si 3.6047E-07 2.6588E-07 5.6946E-07
SiO2 2.5305E-08 1.8665E-08 3.9975E-08
Si2C 6.1848E-09 4.5618E-09 9.7704E-09
Si2 1.5062E-10 1.1110E-10 2.3795E-10
O 1.2484E-11 9.2081E-12 1.9722E-11
SiC 3.3665E-12 2.4831E-12 5.3182E-12
Si3 1.5133E-12 1.1162E-12 2.3907E-12
C 1.4786E-12 1.0906E-12 2.3358E-12
C3 6.4621E-13 4.7664E-13 1.0209E-12
C2 5.5310E-14 4.0796E-14 8.7377E-14
O2 2.7281E-15 2.0122E-15 4.3097E-15
O3 6.0308E-30 4.4483E-30 9.5272E-30
TOTAL: 1.3558E+00 1.0000E+00 1.0000E+00
mol ACTIVITY
SiC 6.6110E-01 1.0000E+00
SiO2(cristobali) 3.2204E-01 1.0000E+00
SiO2(tridymite) 0.0000E+00 9.9918E-01
SiO2(liquid) 0.0000E+00 9.7125E-01
SiO2(quartz) T 0.0000E+00 9.0635E-01
C 0.0000E+00 7.3985E-01
Si 0.0000E+00 4.6901E-02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
2.33923E+02 -3.33851E+05 4.57845E+02 -1.20376E+06 1.00000E+02
Mole fraction of system components:
GAS
C 0.49375
O 0.50003
Si 6.2188E-03
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
! As you can see from the two output tables produced by the calls to ! TQCEL, the only difference is the asterisk that marks the target ! variable (pressure) in the second table. All calculated values are ! otherwise the same. ! Use TQGETR with its option 'VT' to get the calculated total ! volume. Note that TQGETR's option 'VT' is _not_ related to ! TQSETC/TQSTEC's option 'VT'. The latter only specifies that the total ! volume of the system should be _retrieved_, and can always be ! used. The use of TQGETR's option 'VT' need not be changed in any way ! or discontinued with any future version of ChemApp. CALL TQGETR('VT ', 0, 0, RESULT, NOERR) WRITE(*,FMT=10) 'Equilibrium volume (total): ', RESULT, ' dm^3' END
Equilibrium volume (total): 100.00 dm^3
9 Worked examples
In this section, a few 'worked examples' of ChemApp applications will be introduced.
The code examples presented in this section are intended to show how ChemApp is used to solve a specific thermochemical problem, with each example focussing on a different aspect of ChemApp's calculational capabilities. By working through these examples, anyone who is new to ChemApp will get an impression of what ChemApp can be used for, and how, and will hopefully get ideas on how to take the first steps toward solving his or her own problems.
The aim of these programs is to keep the code focussed on the use of the ChemApp subroutines, and to keep everything else simple, in order to not distract from the main issue.
The description of each example is divided into four sections:
| Definition of goals: | |
|---|---|
| states what the purpose of the example program is. | |
| Tools and concepts used: | |
| describes the basic concepts employed in the subsequent implementation, and which ChemApp subroutines are used. | |
| Implementation: | presents the commented source code of the resulting program. |
| Enhancements: | lists suggestions for enhancements to the example that you can add yourself, to gain further understanding of the matter by adding your own code. |
The worked examples presented here are:
- Print data-file contents
- Equilibrium composition of mixture phases
- Phase equilibria and phase target calculations
- A simple process using streams
- One-dimensional phase mapping
9.1 Print data-file contents
The first worked example is a program which reads a thermochemical data-file and prints a table of its contents.
Definition of goals
The aim is to get an overview on what is contained in a particular thermochemical data-file, for instance the ones supplied with this version of ChemApp, or any other data-file that you use with it. It will familiarise the ChemApp user with the concept of how ChemApp handles system components, phases, and phase constituents through index numbers.
The list that the program produces should include information on
- the system components,
- the phases,
- and for each phase the associated model and the list of its phase constituents.
For a definition of the terms used above, see Chapter Definition of terms.
Tools and concepts used
For the program, we will make use of almost all subroutines belonging to group 2 (Identifying Phases, Constituents, and Components). These are
- for system components: TQNOSC, TQINSC, TQGNSC;
- for phases: TQNOP, TQINP, TQGNP, TQMODL;
- and for phase constituents: TQNOPC, TQINPC, TQGNPC, TQPCIS.
The program will first need to determine the number of system components contained in the data-file, then get the name of each of them in a loop. Next, it will do the same for the phases, with the model name displayed as well. It will also print the number of phase constituents, if the phase is a mixture phase. And finally, it will display the names of the phase constituents of each of the mixture phases contained in the data-file, and check each one whether it is permitted to be used as an incoming species. If one or more are not permitted, a note is added to the list.
Implementation
! Print data-file contents PROGRAM CAF100 IMPLICIT NONE INTEGER NOERR, I, J, NSCOM, NPHASE, NPCON, INPCIS, NNPERM CHARACTER FNAME*12, SCNAME*24, PNAME*24, MODEL*6, PCNAME*24 ! Initialise the number of phase constituents which cannot be used as ! incoming species to 0 NNPERM = 0 ! Initialise ChemApp CALL TQINI(NOERR) FNAME = 'cosi.dat' ! For this program to be flexible, add code that asks for the name of ! the data-file instead ! Open the thermochemical data-file cosi.dat (system C-O-Si) C for ! reading CALL TQOPNA(FNAME, 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) WRITE(*,FMT='(1X,A)') 'Contents of data-file ' // FNAME ! *************************************************************** ! Information on system components ! *************************************************************** ! Get the number of system components CALL TQNOSC(NSCOM, NOERR) WRITE(*,FMT='(1X,A,I3)') 'Number of system components: ', * NSCOM ! Print the names of all system components WRITE(*,FMT='(1X,A)') 'List of system components:' DO I=1, NSCOM CALL TQGNSC(I, SCNAME, NOERR) WRITE(*,FMT='(1X,I4,A)') I, ' : ' // SCNAME ENDDO WRITE(*,*)
Contents of data-file cosi.dat Number of system components: 3 List of system components: 1 : C 2 : O 3 : Si
! *************************************************************** ! Information on phases ! *************************************************************** ! Get the number of phases CALL TQNOP(NPHASE, NOERR) WRITE(*,FMT='(1X,A,I3)') 'Number of phases: ', * NPHASE ! Print the names of all phases, the associated model, and the number of ! phase constituents, if it is a mixture phase. WRITE(*,FMT='(1X,A)') 'List of phases:' DO I=1, NPHASE ! Get the phase name CALL TQGNP(I, PNAME, NOERR) ! Get the model name CALL TQMODL(I, MODEL, NOERR) ! Get its number of phase constituents CALL TQNOPC(I, NPCON, NOERR) ! If this phase has more than 1 phase constituent, it is a mixture ! phase... IF (NPCON .GT. 1) THEN WRITE(*,FMT='(1X,I4,A,I4,A)') I, ' : ' // PNAME // ' ' // * MODEL, NPCON, ' constituents' ! ... otherwise it is a stoichiometric compound ELSE WRITE(*,FMT='(1X,I4,A)') I, ' : ' // PNAME // ' ' // MODEL ENDIF ENDDO WRITE(*,*)
Number of phases: 8 List of phases: 1 : GAS IDMX 15 constituents 2 : C PURE 3 : Si PURE 4 : SiC PURE 5 : SiO2(quartz) PURE 6 : SiO2(tridymite) PURE 7 : SiO2(cristobali) PURE 8 : SiO2(liquid) PURE
! *************************************************************** ! Information on phase constituents of mixture phases ! *************************************************************** ! Print information on phase constituents WRITE(*,FMT='(1X,A)') 'List of phase constituents of ' // * 'mixture phases:' DO I=1, NPHASE ! Get its number of phase constituents CALL TQNOPC(I, NPCON, NOERR) ! If this phase has more than 1 phase constituent, it is a mixture ! phase, and we want to print the list of phase constituents IF (NPCON .GT. 1) THEN CALL TQGNP(I, PNAME, NOERR) WRITE(*, FMT='(1X,A)') 'List of phase constituents of ' // * 'phase ' // PNAME ! Get and print the name of each phase constituent of the current phase DO J=1, NPCON CALL TQGNPC(I, J, PCNAME, NOERR) ! Check whether the phase constituent may be used as incoming ! species. If not, increase the total number of such phase constituents ! by 1 and print a mark. CALL TQPCIS(I, J, INPCIS, NOERR) IF (INPCIS .EQ. 0) THEN NNPERM = NNPERM + 1 WRITE(*,FMT='(1X,I4,A)') J, ' : ' // PCNAME // ' X' ELSE WRITE(*,FMT='(1X,I4,A)') J, ' : ' // PCNAME ENDIF ENDDO WRITE(*,*) ENDIF ENDDO ! If there have been phase constituents which cannot be used as incoming ! species, print a note. IF (NNPERM .GT. 0) THEN WRITE(*,FMT='(1X,A, I3,A)') 'Note: ', NNPERM, ' phase ' // * 'constituent(s) marked with "X" is/are not permitted ' WRITE(*,FMT='(1X,A)') ' as incoming species.' ENDIF END
List of phase constituents of mixture phases: List of phase constituents of phase GAS 1 : C 2 : C2 3 : C3 4 : CO 5 : CO2 6 : O 7 : O2 8 : O3 9 : Si 10 : Si2 11 : Si2C 12 : Si3 13 : SiC 14 : SiO 15 : SiO2
Suggestions for enhancements
- Include information on the molecular mass of the system components.
- Include information on the molecular mass and the stoichiometry of the mixture phase constituents and of the stoichiometric compounds.
- For systems which contain mixture phases modeled with sublattices (e.g. Co-Cr-Fe, see SUBL-EX.DAT (System Co-Cr-Fe)), include information on the sublattice details.
9.2 Equilibrium composition of mixture phases
This example will demonstrate what information can be obtained from ChemApp regarding the equilibrium composition of mixture phases. Along the way, the important concept of the stoichiometry matrix (see also Definition of a stoichiometry matrix for details) will be applied. The last calculation will demonstrate the relationship between partial and integral extensive property values for mixture phases, using a non-ideal liquid as an example.
Definition of goals
The aim is to show how ChemApp is used to retrieve information about the equilibrium state of a mixture phase, the gas phase of the system C-O-Si (data-file cosi.dat) being used as an example.
The information to be retrieved includes
- the equilibrium amount of the phase*,
- the equilibrium amounts of the phase constituents*,
- the mole fractions of the phase constituents*,
- the fractions of the system components in the phase, relative to the other system components in the same phase,
- the fractions of the system components in the phase, relative to the same system component in the whole system.
The system Pb-Sn (data-file pbsn.dat) is then used to
- retrieve integral properties of the mixture phase and partial properties of its phase constituents*,
- and show how ChemApp is used to check their relation.
Note that the items marked with an asterisk (*) indicate that the values retrieved for the properties involved are model-dependent. This is potentially always the case when getting information about phase constituents, as different mixture phase models might lead to different phase constituents. For example, when the Redlich-Kister formalism is applied to model a melt, a different set of phase constituents might be used to describe its thermochemical properties than when the quasichemical model is applied to the same melt. Thus also the information and variable values associated with the phase constituents will be different. Even values for variables like the amount of the phase might differ, if the amount is retrieved in mol and the number and/or type of phase constituents differ between two models.
Tools and concepts used
When using mixture phases in equilibrium calculations it is important to familiarise oneself with how the equilibrium state of these phases can be characterised. It is useful to understand which of the results that can be obtained from ChemApp are based on phase-internal information only, and which need phase-external information, relating to the equilibrium states of other phases and the whole system.
It will be shown that the stoichiometry matrix provides important information about the phase constituents, and how it is used to gain some of the results listed above. Familiarising oneself with the concept of the stoichiometry matrix is crucial to anybody who wants to understand the relationship between a mixture phase and its phase constituents. This is especially true for those who plan on assessing mixture phase data themselves, and thus editing thermochemical data-files.
ChemApp also allows for the retrieval of extensive properties of both phases and phase constituents. The partial and integral values for a phase and its constituents are of course related, and it is useful to see how ChemApp can be used to demonstrate this relationship.
Implementation
! Equilibrium composition of mixture phases PROGRAM CAF101 IMPLICIT NONE CHARACTER NAME*24 DOUBLE PRECISION STOI(30), VALS, VALUE, AGAS, ASYS, VALUES(50), * FRACSI, SITOT, WMASS, XSN, HINT1, HINT2, HPB, HSN INTEGER NOERR, NUMCON, ICO, ISIO2, NPCON, I, NSCOM, ISI, * IPB, ISN, ILIQ ! Initialise ChemApp CALL TQINI(NOERR) ! Open the thermochemical data-file cosi.dat (system C-O-Si) ! for reading CALL TQOPNA('cosi.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! For the present example, the input composition consists of 2 mol of ! CO/GAS/ and 3 mol of quartz CALL TQINPC('CO ', 1, ICO, NOERR) CALL TQSETC('IA ', 1, ICO ,2.D0, NUMCON, NOERR) CALL TQINP('SiO2(quartz)', ISIO2, NOERR) CALL TQSETC('IA ', ISIO2, 1, 3.D0, NUMCON, NOERR) ! The temperature at which the equilibrium is to be calculated is set to ! 1500 K CALL TQSETC('T ', 0, 0, 1500.D0, NUMCON, NOERR) ! Calculate the equilibrium and print a ChemSage result table CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 1500 K
P = 1 bar
V = 249.28 dm3
STREAM CONSTITUENTS AMOUNT/mol
CO/GAS/ 2.0000E+00
SiO2(quartz) 3.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
CO 1.9974E+00 9.9935E-01 9.9935E-01
CO2 1.2879E-03 6.4438E-04 6.4438E-04
SiO 1.9134E-06 9.5730E-07 9.5730E-07
SiO2 3.9280E-12 1.9652E-12 1.9652E-12
Si 7.1155E-14 3.5600E-14 3.5600E-14
O 2.6014E-14 1.3015E-14 1.3015E-14
C 3.4466E-17 1.7244E-17 1.7244E-17
O2 2.0479E-17 1.0246E-17 1.0246E-17
C3 8.9044E-18 4.4551E-18 4.4551E-18
C2 2.6909E-19 1.3463E-19 1.3463E-19
Si2C 5.6221E-20 2.8129E-20 2.8129E-20
SiC 9.2988E-21 4.6524E-21 4.6524E-21
Si2 3.6182E-22 1.8103E-22 1.8103E-22
Si3 2.2100E-28 1.1057E-28 1.1057E-28
O3 1.8757E-34 9.3844E-35 9.3844E-35
TOTAL: 1.9987E+00 1.0000E+00 1.0000E+00
mol ACTIVITY
SiO2(tridymite) 3.0000E+00 1.0000E+00
C 1.2860E-03 1.0000E+00
SiO2(cristobali) 0.0000E+00 9.9878E-01
SiO2(quartz) 0.0000E+00 9.4548E-01
SiO2(liquid) 0.0000E+00 8.2521E-01
SiC 0.0000E+00 5.2057E-04
Si 0.0000E+00 3.5247E-06
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
2.90050E+02 -2.62627E+06 9.37547E+02 -4.03259E+06 2.49275E+02
Mole fraction of system components:
GAS
C 0.49984
O 0.50016
Si 4.7850E-07
! First, get the equilibrium amount of the gas phase, which is the only ! mixture phase in the system. Note that since we did not change any ! units, the default amount unit (mol) is used. CALL TQGETR('A ', 1, 0, AGAS, NOERR) WRITE(*,FMT='(A,G12.5)') 'Equilibrium amount of gas phase ' // * 'in mol: ', AGAS
Equilibrium amount of gas phase in mol: 1.9987
! To determine the mole fraction of the gas phase, relative to the whole ! system, we first need to get the amount contained in the system CALL TQGETR('A ', 0, 0, ASYS, NOERR) WRITE(*,FMT='(A,G12.5)') 'Mole fraction of the gas phase: ', * AGAS/ASYS
Mole fraction of the gas phase: 0.39974
! The equilibrium amounts of all phase constituents of the gas phase can ! be obtained by a call to TQGETR which passes an array together with ! the proper INDEX/INDEXP combination CALL TQGETR('A ', 1, -1, VALUES, NOERR) ! Print the equilibrium amount of each phase constituent CALL TQNOPC(1, NPCON, NOERR) WRITE(*,*) 'Equilibrium amounts of gas phase constituents' DO I=1, NPCON CALL TQGNPC(1, I, NAME, NOERR) WRITE(*,FMT='(I4,A,G12.5,A)') I, ' : ' // NAME, * VALUES(I), ' mol' ENDDO
Equilibrium amounts of gas phase constituents 1 : C 0.34466E-16 mol 2 : C2 0.26909E-18 mol 3 : C3 0.89044E-17 mol 4 : CO 1.9974 mol 5 : CO2 0.12879E-02 mol 6 : O 0.26014E-13 mol 7 : O2 0.20479E-16 mol 8 : O3 0.18757E-33 mol 9 : Si 0.71155E-13 mol 10 : Si2 0.36182E-21 mol 11 : Si2C 0.56221E-19 mol 12 : Si3 0.22100E-27 mol 13 : SiC 0.92988E-20 mol 14 : SiO 0.19134E-05 mol 15 : SiO2 0.39280E-11 mol
! The mole fractions of the phase constituents can be calculated using ! the values just retrieved ! Print the mole fractions of each phase constituent WRITE(*,*) 'Mole fractions of gas phase constituents' DO I=1, NPCON CALL TQGNPC(1, I, NAME, NOERR) WRITE(*,FMT='(I4,A,G12.5)') I, ' : ' // NAME, * VALUES(I)/AGAS ENDDO
Mole fractions of gas phase constituents 1 : C 0.17244E-16 2 : C2 0.13463E-18 3 : C3 0.44551E-17 4 : CO 0.99935 5 : CO2 0.64438E-03 6 : O 0.13015E-13 7 : O2 0.10246E-16 8 : O3 0.93844E-34 9 : Si 0.35600E-13 10 : Si2 0.18103E-21 11 : Si2C 0.28129E-19 12 : Si3 0.11057E-27 13 : SiC 0.46524E-20 14 : SiO 0.95730E-06 15 : SiO2 0.19652E-11
! Information relating the system components (in the present case C, O, ! and Si) to the phases can also be obtained. ! Get the mole fraction of the system components in the gas phase CALL TQNOSC(NSCOM, NOERR) WRITE(*,*) 'Mole fractions of system components in the gas phase' DO I=1, NSCOM CALL TQGNSC(I, NAME, NOERR) CALL TQGETR('XP ', 1, I, VALUE, NOERR) WRITE(*,FMT='(I4,A,G12.5)') I, ' : ' // NAME, * VALUE ENDDO
Mole fractions of system components in the gas phase 1 : C 0.49984 2 : O 0.50016 3 : Si 0.47850E-06
! To get the fraction of a system component in the gas phase, we have to ! make use of the information contained in the stoichiometry matrix. CALL TQINSC('Si ', ISI, NOERR) FRACSI = 0.D0 ! Get the equilibrium amount of each gas phase constituent, then use the ! stoichiometry matrix returned by TQSTPC to determine how much Si is ! contained in the constituent, and sum it all up DO I = 1, NPCON CALL TQSTPC(1, I, STOI, WMASS, NOERR) CALL TQGETR('A ', 1, I, VALUE, NOERR) FRACSI = FRACSI + STOI(ISI)*VALUE ENDDO ! Get the total amount of Si in the system CALL TQGETR('a ',0,ISI,SITOT,NOERR) WRITE(*,FMT='(A,G12.5)') 'Mole fraction of system component ' // * 'Si in the gas phase: ', FRACSI/SITOT
Mole fraction of system component Si in the gas phase: 0.63779E-06
! To demonstrate how ChemApp can be used to check the relationship ! between partial and integral values of extensive properties, a ! different data-file for a system that contains non-ideal mixture ! phases is used. ! Open a new thermochemical data-file (pbsn.dat, system Pb-Sn) ! for reading CALL TQOPNA('pbsn.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Find out the system component index numbers for Pb and Sn, as well as ! the phase index number for the liquid phase: CALL TQINSC('Pb ', IPB, NOERR) CALL TQINSC('Sn ', ISN, NOERR) CALL TQINP('LIQUID ', ILIQ, NOERR) ! Using the index numbers just determined, we can enter our incoming ! amounts, in this case an alloy consisting of 0.7 mol Sn and 0.3 mol Pb: XSN = 0.7D0 CALL TQSETC('IA ', 0, ISN, XSN, NUMCON, NOERR) CALL TQSETC('IA ', 0, IPB,1.D0 - XSN, NUMCON, NOERR) ! Set the temperature to 600 K. Under these conditions, the liquid phase ! is the only stable phase in this system (see Figure ref(pbsn2_pd)). CALL TQSETC('T ', 0, 0, 600.D0, NUMCON, NOERR) ! Calculate the equilibrium CALL TQCE(' ', 0, 0, VALS, NOERR) ! Get the integral enthalpy of the liquid phase CALL TQGETR('H ', ILIQ, 0, HINT1, NOERR) ! Get the partial molar enthalpies of Sn/LIQUID/ and Pb/LIQUID/ CALL TQGETR('HM ', ILIQ, ISN, HSN, NOERR) CALL TQGETR('HM ', ILIQ, IPB, HPB, NOERR) ! Calculate the integral enthalpy of the liquid phase from the two ! partial values HINT2 = XSN * HSN + (1.D0 - XSN) * HPB ! Print both values WRITE(*,FMT='(A,F12.3,A)') 'HINT1 = ', HINT1,' J' WRITE(*,FMT='(A,F12.3,A)') 'HINT2 = ', HINT2,' J'
HINT1 = 16066.466 J HINT2 = 16066.466 J
! As expected, the two values are the same END
Suggestions for enhancements
- Before adding to the code, answer the following questions for yourself:
- In the above example, the fraction of system component Si in the gas phase has been calculated twice. Since the values are different, what Si fraction exactly has been calculated in each case?
- Which of the results obtained using the calls to TQGETR can also be read from the ChemSage/ChemApp result table printed by TQCEL?
- Looking at the ChemSage/ChemApp result table, why does the column labelled 'fugacity' contain the same values as the column labelled 'mole fraction'?
- When retrieving the integral enthalpy for the Pb-Sn melt, the 'H' option of TQGETR is used. Will it make a difference for the example used if the molar enthalpy (option 'HM') is used instead?
- What happens when a different total amount in mol is entered into the equilibrium calculation instead, will the options 'H' and 'HM', as used in the example, still result in both integral enthalpies to be equal?
- Extend the part of the program which gets the fraction of the system component Si in the gas phase (relative to the total amount of Si in the whole system) so that a list of fractions for all system components is printed.
- Calculate the integral Gibbs energy of the Pb-Sn melt without using the options 'G' or 'GM' for TQGETR. Verify it in a similar fashion as it is done for the enthalpy in the above example.
9.3 Phase equilibria and phase target calculations
The target calculations ChemApp is able to perform are a very powerful tool to find one's way around a phase diagram. The following program demonstrates, for the binary system Pb-Sn (see PBSN.DAT (System Pb-Sn)), how target calculations and standard equilibrium calculations can be used in phase constitution calculations. Figure The Pb-Sn phase diagram shows the binary phase diagram Pb-Sn. The items labeled will be calculated in the course of this application example.
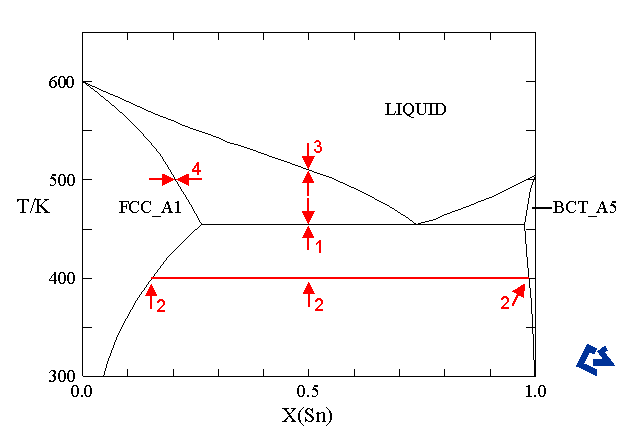
The Pb-Sn phase diagram
Definition of goals
The type of information to be obtained should include the following:
- The determination of temperatures at which a phase (e.g. the liquid) becomes stable for an alloy of a given composition (formation phase target calculation, item 1 in Figure The Pb-Sn phase diagram)
- Retrieving the necessary mole fractions and amounts which are associated with the 'lever rule' for an alloy at a given composition and temperature (item 2 in Figure The Pb-Sn phase diagram).
- The determination of the temperature at which a phase (e.g. a solid phase from the liquid) starts to precipitate (precipitation target calculations, item 3 in Figure The Pb-Sn phase diagram).
- Calculations similar to the ones above, but with composition instead of temperature as the target variable (item 4 in Figure The Pb-Sn phase diagram).
Tools and concepts used
Target calculations are a very powerful concept in ChemApp. To perform them, both the target itself needs to be specified (using TQSETC or TQSTEC), as well as the target variable (using TQCE/TQCEL). The result is that ChemApp then performs a range of equilibrium calculations, varying the target variable until the target is met. ChemApp allows for the calculation of phase targets and extensive property targets. Applications of the former are demonstrated in this program, an example for the latter would be the calculation of an adiabatic temperature (e.g. the 'flame temperature' in combustion calculations, as demonstrated in the example for TQCE).
As for the type of target, phase target calculations come in two basic 'flavours'. The first one, a formation phase target calculation, tells ChemApp to look for the condition under which a specific phase forms (usually, but not necessarily with the amount of that phase being zero). The other one, a precipitation target calculation, is defined as one where the target phase is the only one formed at equilibrium, and the second most stable phase has unit activity. Note that precipitation target calculations can only be performed for solution phases.
As for the target variables, temperature, pressure, volume, and composition (see also Table State variables for defining targets when calling TQCE/TQCEL) are possible. In the present program, the use of temperature and composition will be demonstrated.
Implementation
! Phase equilibria and phase target calculations ! Note that this program contains phase target calculations, ! which cannot be performed with the 'light' version of ChemApp PROGRAM CAF102 IMPLICIT NONE CHARACTER NAME*24, MODEL*6 DOUBLE PRECISION VALUE, VALS(2), FSNBCT, FSNFCC, * ABCT, AFCC, EPS INTEGER NOERR, ISLITE, NPHASE, NUMCON, I, NPCON, * IPB, ISN, I_LIQ, I_BCT, I_FCC ! Initialise ChemApp CALL TQINI(NOERR) ! Open the thermochemical data-file pbsn.dat (system Pb-Sn) ! for reading CALL TQOPNA('pbsn.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! To find out about the phases contained in the data-file, print out the ! names of all phases and the associated models CALL TQNOP(NPHASE, NOERR) DO I=1, NPHASE CALL TQGNP(I, NAME, NOERR) CALL TQMODL(I, MODEL, NOERR) WRITE(*,FMT='(I2,A)') I, ': ' // NAME // MODEL ENDDO
1: LIQUID RKMP 2: BCT_A5#1 RKMP 3: BCT_A5#2 RKMP 4: FCC_A1 RKMP
! As can be seen from the short table printed above, all phases present ! in the data-file are mixture phases, since the model identifier ! 'PURE' does not appear anywhere. Also, the phase BCT_A5 is entered ! twice into the data-file. This is required because of the inherent ! metastable miscibility gap in the system. ! **************************************************************** ! Formation phase target calculation ! Item 1 in the Pb-Sn phase diagram ! **************************************************************** ! The first calculation will be the determination of the eutectic ! temperature. To do this, we need to define an alloy composition which, ! upon solidification, would undergo a eutectic transformation. An alloy ! consisting of 50% Sn and 50% Pb is a good candidate. We will tell ! ChemApp to use an alloy of this composition, and ask for the ! temperature (which thus is the target variable) at which the liquid ! phase first becomes stable (which thus is the target). Applying the ! 'formation phase target liquid' thus means that ChemApp will determine ! the point where the liquid phase is stable (activity is unity) and its ! equilibrium amount is the one selected by the programmer (in our case ! zero). ! We will use 'global conditions', because we are not interested in ! calculating extensive property (e.g. 'heat') balances. ! We will use the system components to input our incoming amounts. Note ! that it does not matter whether we input our incoming amounts as ! system components, or through any of the mixture phase constituents or ! stoichiometric compounds, since we are not interested in the extensive ! property balances. ! Find out the system component index numbers for Pb and Sn: CALL TQINSC('Pb ', IPB, NOERR) CALL TQINSC('Sn ', ISN, NOERR) ! Using the index numbers just determined, we can enter our incoming ! amounts: ! Entering 0.5 mol Pb CALL TQSETC('IA ', 0, IPB, 0.5D0, NUMCON, NOERR) ! Entering 0.5 mol Sn CALL TQSETC('IA ', 0, ISN, 0.5D0, NUMCON, NOERR) ! Check if we are working with the 'light' version. ! If we do, omit the following phase target calculation(s). CALL TQLITE(ISLITE, NOERR) IF (ISLITE .EQ. 1) THEN WRITE(*,FMT='(3(1X,A,/))') * '*** Phase target calculations have been omitted here,', * '*** since they are not possible with the', * '*** ''light'' version of ChemApp.' ELSE ! The 'LIQUID' phase is the target phase. From the first output we see ! that it has the index number 1, which can also be determined by a call ! of TQINP: CALL TQINP('LIQUID ', I_LIQ, NOERR) ! Using TQSETC, we define the _target_: formation phase target 'LIQUID', ! zero amount CALL TQSETC('A ', I_LIQ, 0, 0.D0, NUMCON, NOERR) ! The _target variable_ is passed to TQCE/TQCEL: The target variable ! used is the temperature, with 300 K as the first guess. This estimate ! does not need to be very accurate. VALS(1) = 300.0 CALL TQCE('T ', 0, 0, VALS, NOERR) ! Retrieve the calculated temperature CALL TQGETR('T ', 0, 0, VALUE, NOERR) WRITE(*,FMT='(1X,A,G12.5,A)') 'The eutectic temperature is ', * VALUE, ' K'
The eutectic temperature is 454.56 K
ENDIF
! ****************************************************************
! Phase equilibrium calculation (verification of the 'lever rule')
! Item 2 in the Pb-Sn phase diagram
! ****************************************************************
! Now, for a temperature below the eutectic one, calculate the mole
! fraction of Sn in both eutectic phases (FCC and BCT_A5#1)
! The alloy composition is left unchanged (50% Sn, 50% Pb), but now the
! temperature is set to 400 K
CALL TQSETC('T ', 0, 0, 400.D0, NUMCON, NOERR)
! Calculate the equilibrium
CALL TQCE(' ', 0, 0, VALS, NOERR)
! To check the lever rule for this case, and compare it to the graphical
! representation in the Pb-Sn phase diagram, we need the mole fraction
! of Sn in the BCT_A5#1 and FCC phases (FSNBCT and FSNFCC), the amounts
! of BCC and FCC phase (ABCT and AFCC), plus the mole fraction of Sn in
! the alloy, which is 0.5.
! Get the equilibrium amount of phase BCT_A5#1 in mol
CALL TQINP('BCT_A5#1 ', I_BCT, NOERR)
CALL TQGETR('A ', I_BCT, 0, ABCT, NOERR)
! Get the mole fraction of Sn in BCT_A5#1
CALL TQGETR('XP ', I_BCT, ISN, FSNBCT, NOERR)
! Get the equilibrium amount of phase FCC in mol
CALL TQINP('FCC ', I_FCC, NOERR)
CALL TQGETR('A ', I_FCC, 0, AFCC, NOERR)
! Get the mole fraction of Sn in FCC
CALL TQGETR('XP ', I_FCC, ISN, FSNFCC, NOERR)
! For the lever rule condition to be satisfied, the following equation
! has to hold: (0.5 - FSNFCC)*AFCC - (FSNBCT - 0.5)*ABCT = EPS
! with EPS being zero within the numerical precision.
EPS = (0.5D0 - FSNFCC)*AFCC - (FSNBCT - 0.5D0) * ABCT
WRITE(*,FMT='(1X,A,G12.5)') 'The value of EPS is ', EPS
The value of EPS is -0.27756E-16
! As you can see, the lever rule is sufficiently satisfied. ! Check if we are working with the 'light' version. ! If we do, omit the following phase target calculation(s). CALL TQLITE(ISLITE, NOERR) IF (ISLITE .EQ. 1) THEN WRITE(*,FMT='(3(1X,A,/))') * '*** Phase target calculations have been omitted here,', * '*** since they are not possible with the', * '*** ''light'' version of ChemApp.' ELSE ! **************************************************************** ! Precipitation target calculation, with temperature as target ! variable ! Item 3 in the Pb-Sn phase diagram ! **************************************************************** ! The next calculation is again a target calculation. This time, a ! 'precipitation phase target' is used to determine a point on the ! liquidus curve, the alloy composition being the same as before. Using ! a precipitation phase target tells ChemApp to search for the condition ! under which a second phase becomes stable. ! The subsequent call to TQSETC tells ChemApp to set the following ! _target_: The liquid phase is stable, and a second phase (which does ! not need to be specified) has unit activity. CALL TQSETC('A ', I_LIQ, 0, -1.D0, NUMCON, NOERR) ! Use the temperature as _target variable_ and calculate the equilibrium VALS(1) = 1000.0 CALL TQCE('T ', 0, 0, VALS, NOERR) ! Retrieve the calculated liquidus temperature CALL TQGETR('T ', 0, 0, VALUE, NOERR) WRITE(*,FMT='(1X,A,G12.5,A)') 'The liquidus temperature is ', * VALUE, ' K'
The liquidus temperature is 510.45 K
! **************************************************************** ! Precipitation target calculation, with composition as target ! variable ! Item 4 in the Pb-Sn phase diagram ! **************************************************************** ! A target variable which is especially useful in T-x diagrams with ! rather steep phase boundaries is the composition of an alloy. The ! following example determines the point of the FCC phase boundary at ! 500 K where LIQUID is formed (or precipitates). ! First, for convenience, remove _all_ previously set conditions. This ! is especially useful since we have to remove the conditions describing ! the fixed composition of the alloy (set toward the beginning of the ! program) anyway, as the composition is now the target variable and ! thus supposed to vary. CALL TQREMC(-2, NOERR) ! Set the temperature to 500 K CALL TQSETC('T ', 0, 0, 500.D0, NUMCON, NOERR) ! Set the target: The FCC phase is stable, and a second phase has unit ! activity. CALL TQSETC('A ', I_FCC, 0, -1.D0, NUMCON, NOERR) ! If the composition is the target variable and the total input amount ! should always be 1 mol, we need to vary two compositions ! symmetrically. In the present case, we tell ChemApp to vary the input ! amount of Sn/FCC/ between 0 and 1 to find the target, and to vary ! Pb/FCC/ symmetrically between 1 and 0. ! The upper and lower limits of the composition target variables are ! passed via the array VALS to TQCE. First we pass the limits for ! Sn/FCC/. VALS(1) = 0.0 VALS(2) = 1.0 ! We need to tell ChemApp that we are not finished with our input, since ! we still need to specify the symmetrical limits for Pb/FCC/ ! later. This is done by calling TQCE with the option 'IA0', which tells ! ChemApp not to perform an equilibrium calculation, but to expect more ! incoming amounts. CALL TQCE('IA0 ', I_FCC, ISN, VALS, NOERR) ! Enter the symmetrical limits for Pb/FCC/ VALS(1) = 1.0 - VALS(1) VALS(2) = 1.0 - VALS(2) ! With the next call to TQCE, the information ChemApp needs to perform ! a composition target is complete, so we call ChemApp with the option ! 'IA', upon which the equilibrium is calculated. CALL TQCE('IA ', I_FCC, IPB, VALS, NOERR) ! ChemApp has now varied the composition of the alloy until the desired ! point of the phase boundary has been found. The desired value (the ! mole fraction of Sn in the FCC phase) is retrieved. CALL TQGETR('XP ', I_FCC, ISN, FSNFCC, NOERR) WRITE(*,FMT='(1X,A,G12.5)') 'The mole fraction of Sn is ', FSNFCC ENDIF END
The mole fraction of Sn is 0.20698
Suggestions for enhancements
- Calculate the following elements from the Pb-Sn phase diagram:
- Determine which phase precipitates from the liquid at the point on the liquidus curve that has been calculated as item 3 in the Pb-Sn phase diagram.
- The liquidus line. Make sure that the eutectic point is included (hint: a separate, different target calculation is required for this purpose).
- The FCC phase boundary. Make sure that the end point of the eutectic line (part of the 3-phase equilibrium FCC+LIQUID+BCT_A5) is included.
- The zero phase fraction (ZPF) line for BCT_A5. If you are unfamiliar with the concept of ZPF lines, see for instance the The SGTE Casebook - Thermodynamics at work [Hac96], or the FactSage-Teach handbook.
- At T = 475 K, calculate the activities of the phases FCC, LIQUID, and BCT_A5 between xSn = 0.0 and 1.0, and compare the results with the phase regions in the phase diagram.
- At various temperatures (e.g. T = 650 K, 550 K, 475 K, 400 K, and the eutectic temperature), generate plots of the integral Gibbs energies of the phases FCC, LIQUID, and BCT_A5 between xSn = 0.0 and 1.0 using your favourite plotting program, and verify that the information they provide about the phase equilibria at these temperatures is consistent with the phase diagram shown above.
9.4 A simple process using streams
ChemApp can be used as a very powerful tool at the heart of a process simulation program to determine the chemical equilibria and heat balances involved. The concept of streams in ChemApp is especially helpful in this respect. A stream (see also Setting initial conditions) is a mixture of non-reacted matter of constant temperature and pressure which is transferred to a reaction zone. Several such streams can be defined to constitute the material input for an equilibrium calculation.
It is easy to see how, using the concept of streams, ChemApp can be used to model complex processes and to supplement existing flowsheet programs and process simulators through its ability to calculate highly complex multicomponent, multiphase equilibria very quickly and reliably.
In the present example, a combustion process is simulated using the system C-N-O. While the system itself is very simple, it provides a good basis for demonstration. Particular attention will be paid to the conditions leading to the formation of condensed phases (i.e. precipitates formed during the combustion). With the simple data-file used, this possible precipitate is solid carbon.
In real-life applications similar to the one below, ChemApp may be used with data-files describing systems of up to 30 system components containing many hundreds of species, investigating for instance the cooling of exhaust gases from incineration processes. Using multiple connected reaction 'chambers', each at a different temperature, the formation of condensed phases in each stage can be analysed. The result is a simulation program which permits the understanding and optimisation of the process described.
The process we will look at can be sketched as shown in Figure Sketch of the combustion process investigated.
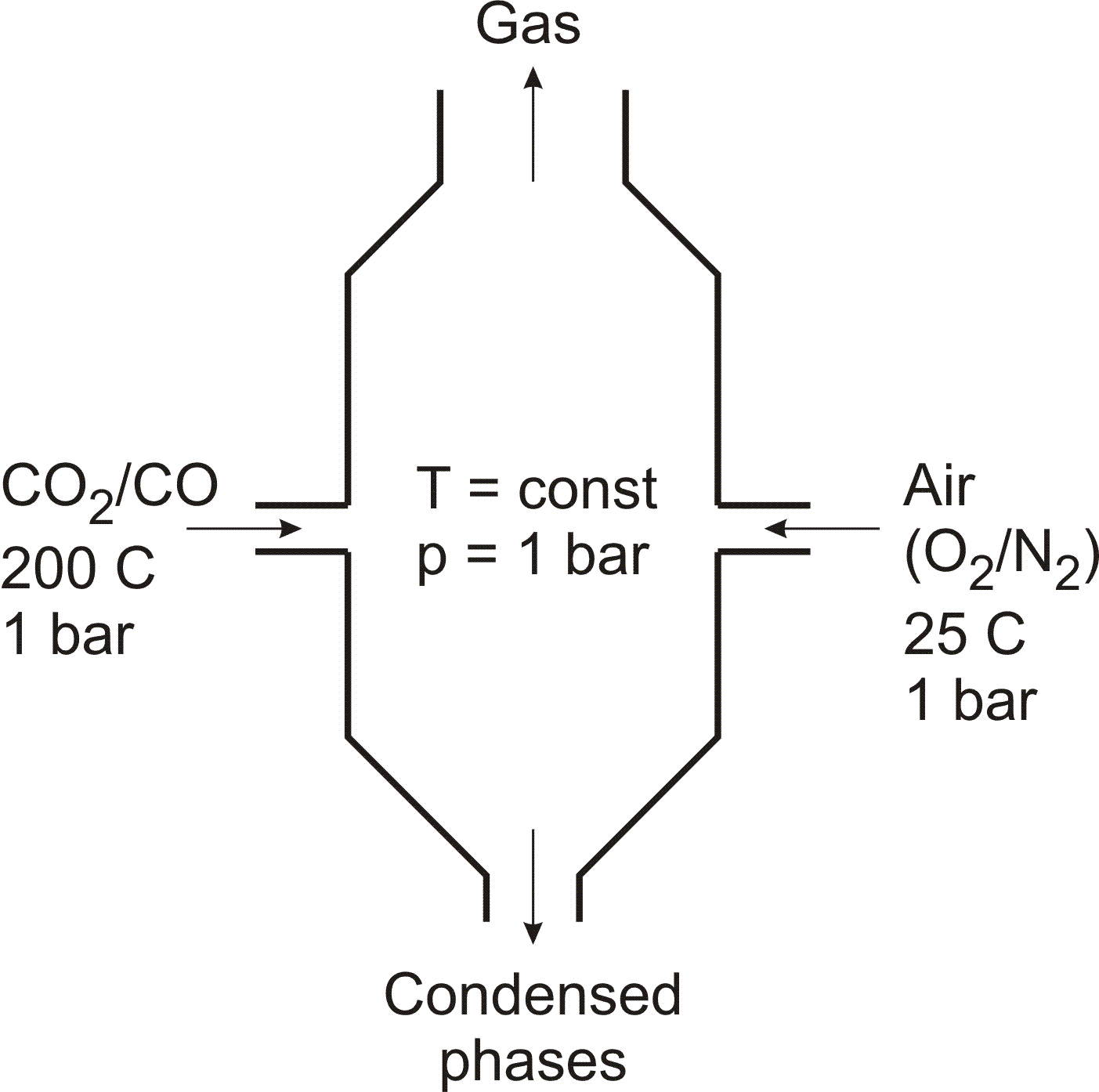
Sketch of the combustion process investigated
In this example we assume that a mixture of CO2 and CO at 200 C and 1 bar, which can be thought of as an exhaust gas from a previous incomplete combustion process, is entering a reactor. To further combust this gas, air at room temperature is supplied. The air in this example is supposed to consist of O2 and N2 at a ratio of 1:4. The amount of air supplied will be kept constant at first, but varied later on to investigate its influence on the formation of condensed phases. The reaction chamber is kept at a constant temperature. This will also be varied later to look at its influence on the process. In the process considered, the material formed at equilibrium would then be separated into a gas phase and the condensed phases, which leave the reactor in different streams. If the model would be extended to reflect a more complex process, consisting of several different units, these two outputs could in turn make up input streams for subsequent reactor units.
Definition of goals
- At first, a single equilibrium calculation is performed for a specific amount of air supplied and for one reaction temperature. The calculated equilibrium state is then investigated, with special attention paid to the formation of phases and some selected extensive property values (heat and volume balances).
- The reaction temperature and the amount of air supplied are then varied separately to investigate their influence on the amount of condensed phases formed.
- To get a feeling for their combined effect, both parameters are then varied simultaneously, and are displayed in a '3D' surface and contour diagram together with the amount of condensed phase formed.
- Assuming that one is particularly interested in finding the conditions under which no precipitates are formed in the process, the necessary values for the reaction temperature and the amount of air supplied to achieve this condition are determined.
Tools and concepts used
As mentioned above, the concept of streams provides the basis for the simulation of the process. By using streams instead of global conditions, it is not only possible to calculate the chemical equilibrium of the process considered, but to also determine the extensive property balances (see also the notes in Extensive properties of streams for more information on how the extensive properties are calculated). The concept of streams makes it also easier to write simulation code for a process in which several individual paths of material flow need to be considered. Translation of such a 'flowsheet' setup into a set of streams is straightforward.
When using streams, the most important ChemApp subroutines are TQSTTP (used to define a stream and assign it a name, temperature, and pressure) and TQSTCA (used to set the constituent amounts for a stream). TQSTEC is then used to set conditions (in the present example, the reaction temperature) for the equilibrium calculation. To retrieve values for the extensive properties of the streams, TQSTXP is called. Another subroutine which is used in connection with streams is TQSTRM, which removes a previously defined stream. It is not called in this example, as it is not necessary to drop any stream from the present simulation. The number of streams (2 in this example) does not change during the course of the simulation.
The last goal stated in the list above requires also the use of target calculations, since we will be looking for a specific condition (the formation of condensed phases) to be met, and we want ChemApp to tell us at which values of a particular input parameter (which thus becomes the target variable) this happens.
Note that visualising any results from ChemApp in form of graphs requires the use of a separate plotting program or graphics library, since ChemApp itself does not provide any subroutines for graphical output.
Implementation
! A simple process using streams ! Note that this program contains phase target calculations, ! which cannot be performed with the 'light' version of ChemApp ! The following function calculates the sum of all equilibrium amounts ! of the condensed phases. It starts with phase #2, since the gas phase ! is #1. The index number of the last phase to be considered is passed ! as an argument to the function, and is retrieved in the main program ! with a call to TQNOP. DOUBLE PRECISION FUNCTION GETACP(LASTPH) INTEGER LASTPH, I DOUBLE PRECISION AMOUNT GETACP= 0.D0 ! Add up the equilibrium amounts of all condensed phases DO I=2, LASTPH CALL TQGETR('A ', I, 0, AMOUNT, NOERR) GETACP = GETACP + AMOUNT ENDDO END ! Main program PROGRAM CAF103 IMPLICIT NONE INTEGER NOERR, ISLITE, NPHASE, J, K, ICO2, ICO, IO2, IN2 DOUBLE PRECISION VALS(2), TP(2), RESULT, ACOND, AGAS, TEMP, * IAO2, IAN2, GETACP ! Initialise ChemApp CALL TQINI(NOERR) ! Open data-file for reading. In this case, a data-file containing the ! thermochemical data for the system carbon-nitrogen-oxygen is selected. CALL TQOPNA('cno.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Since we will perform an extensive property target calculation later, ! we will use streams. This way we can assign an initial state ! regarding temperature and pressure to the incoming substances, which ! will be taken into account when ChemApp calculates the extensive ! property (e.g. heat) balance. ! Since we are going to use degrees Celsius as temperature unit ! throughout the calculations, we will change the unit now CALL TQCSU('Temperature ', 'C ', NOERR) ! According to the process scheme shown above, define the first stream. ! It is the incoming stream of mixed CO2 and CO at 200 C and 1 bar, ! which we will call 'CO2CO'. The amount of CO2 and CO in this stream is ! set to 0.5 and 2.5 mol, respectively. TP(1) = 200.D0 TP(2) = 1.D0 CALL TQSTTP('CO2CO ', TP, NOERR) ! Set the constituent amounts of CO2 and CO. To do this, get the index ! number of these constituents of the gas phase, which is phase number ! 1. CALL TQINPC('CO2 ', 1, ICO2, NOERR) CALL TQINPC('CO ', 1, ICO, NOERR) ! Use the index numbers just obtained to set the constituent amount of ! CO2 in the stream 'CO2CO' to 0.5 mol. CALL TQSTCA('CO2CO ', 1, ICO2, .5D0, NOERR) ! Similarly, set the amount of CO in the same stream to 2.5 mol. CALL TQSTCA('CO2CO ', 1, ICO, 2.5D0, NOERR) ! Define the second stream ('O2N2'), which is air supplied at room ! temperature and 1 bar for the combustion. The air is assumed to ! consist of O2 and N2 at a ratio of 1:4. The amount of O2 is initially ! set to 0.5 mol (thus N2 = 2.0 mol), and will later be varied. TP(1) = 25.D0 TP(2) = 1.D0 CALL TQSTTP('O2N2 ', TP, NOERR) ! Get the index numbers of O2 and N2. CALL TQINPC('O2 ', 1, IO2, NOERR) CALL TQINPC('N2 ', 1, IN2, NOERR) ! Set the amount of O2 CALL TQSTCA('O2N2 ', 1, IO2, .5D0, NOERR) ! Set the amount of N2 CALL TQSTCA('O2N2 ', 1, IN2, 2.0D0, NOERR) ! Set the temperature of the reactor at which the equilibrium reaction ! (i.e. the combustion) will take place. It is initially set to 600 C at ! first, but will be varied later. CALL TQSTEC('T ', 0, 600.D0, NOERR) ! In the result table to be printed out subsequently, the amount unit ! for the condensed phases formed should be 'gram', while the input was ! given in 'mol'. CALL TQCSU('Amount ', 'gram ', NOERR) ! Calculate the equilibrium and print out the result table for a quick ! overview on the equilibrium state. CALL TQCEL(' ', 0, 0, VALS, NOERR)
T = 600 C
P = 1 bar
V = 340.05 dm3
STREAM CONSTITUENTS AMOUNT/gram TEMPERATURE/C PRESSURE/bar STREAM
CO2/GAS/ 2.2005E+01 200.00 1.0000E+00 1
CO/GAS/ 7.0026E+01 200.00 1.0000E+00 1
O2/GAS/ 1.5999E+01 25.00 1.0000E+00 2
N2/GAS/ 5.6027E+01 25.00 1.0000E+00 2
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
N2 2.0000E+00 4.2699E-01 4.2699E-01
CO2 1.8160E+00 3.8771E-01 3.8771E-01
CO 8.6792E-01 1.8530E-01 1.8530E-01
NO 4.3199E-17 9.2227E-18 9.2227E-18
CNO_NCO 2.6452E-20 5.6473E-21 5.6473E-21
N2O 3.0560E-21 6.5244E-22 6.5244E-22
CN 2.9723E-21 6.3457E-22 6.3457E-22
CNO 2.2356E-21 4.7729E-22 4.7729E-22
C2O 3.9875E-23 8.5130E-24 8.5130E-24
O 8.1188E-24 1.7333E-24 1.7333E-24
O2 3.8588E-24 8.2383E-25 8.2383E-25
N 2.2022E-25 4.7016E-26 4.7016E-26
CN2_NCN 1.0083E-28 2.1527E-29 2.1527E-29
NO2 1.2777E-29 2.7278E-30 2.7278E-30
N3 4.1859E-30 8.9367E-31 8.9367E-31
C2N_CNC 2.0615E-34 4.4012E-35 4.4012E-35
C 8.4509E-35 1.8042E-35 1.8042E-35
CN2_CNN 1.9931E-36 4.2552E-37 4.2552E-37
C3 1.1284E-38 2.4090E-39 2.4090E-39
C2 1.0339E-39 2.2074E-40 2.2074E-40
C2N_CCN 2.5088E-41 5.3561E-42 5.3561E-42
O3 2.8768E-48 6.1417E-49 6.1417E-49
NO3 1.4801E-48 3.1600E-49 3.1600E-49
C4 1.1746E-49 2.5077E-50 2.5077E-50
C5 8.2283E-50 1.7567E-50 1.7567E-50
N2O3 2.7228E-51 5.8129E-52 5.8129E-52
N2O4 9.8675E-65 2.1067E-65 2.1067E-65
N2O5 NOT CALCD. NOT CALCD. <1.0000E-75
TOTAL: 4.6840E+00 1.0000E+00 1.0000E+00
gram ACTIVITY
C 3.7960E+00 1.0000E+00
C_DIAMOND_A4 0.0000E+00 4.7889E-01
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
9.65942E+02 -2.52713E+05 1.89546E+01 -8.16760E+05 1.60052E+02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
1.13495E+03 -7.09401E+05 1.17061E+03 -1.73152E+06 3.40047E+02
Mass fraction of system components:
GAS
C 0.20115
N 0.34960
O 0.44925
! As can be seen from the above table, both the gas phase and carbon are ! stable under the conditions chosen. It can also be seen that N2, CO2, ! and CO are the only majority gas phase species. All other gas phase ! species are produced in negligible amounts only. ! The values for the amounts of condensed phases and the gas phase can ! be retrieved using TQGETR. Since the gas phase, if present in the ! data-file, has always the phase index number 1, this means that we ! have to add up all the amounts of the phases from phase #2 to the last ! one in order to retrieve the total amount of condensed phases. ! Find out how many phases we have in the data-file, and use this number ! in all subsequent calls to GETACP CALL TQNOP(NPHASE, NOERR) ! Get the total equilibrium amount of the condensed phases, using the ! function GETACP declared above. ACOND = GETACP(NPHASE) 11 FORMAT(1X,A,G12.5,A) WRITE(UNIT=*,FMT=11) 'Amount of condensed material: ', * ACOND, ' gram'
Amount of condensed material: 3.7960 gram
! Retrieve the amount of gas phase. Note: The current amount unit is ! still 'gram'. If the amount of the gas phase is desired in 'mol' ! instead, TQCSU would need to be called again. CALL TQGETR('A ', 1, 0, AGAS, NOERR) WRITE(UNIT=*,FMT=11) 'Amount of gas phase: ', AGAS, ' gram'
Amount of gas phase: 160.26 gram
! Retrieve values for some extensive properties. Since we used streams ! for this example, these extensive property values are calculated ! relative to the incoming streams, when the whole system is selected. ! Get the enthalpy change associated with this reaction. CALL TQGETR('H ', 0, 0, RESULT, NOERR) WRITE(UNIT=*,FMT=11) 'Delta H: ', RESULT, ' J' ! The negative sign of this value indicates that energy is released by ! the system. ! Get the change in volume of the process, which is the volume of the ! equilibrium gas phase minus the sum of the volumes of the incoming ! streams. CALL TQGETR('V ', 0, 0, RESULT, NOERR) WRITE(UNIT=*,FMT=11) 'Delta V:', RESULT, ' dm^3' ! Note: using the option 'V' with TQGETR normally would retrieve the ! _total_ volume of the system. There is one exception, in which case it ! retrieves the _change_ in volume, and that is the case when streams ! are used _and_ the whole system (i.e. TQGETR is called using '0,0', as ! above) is selected. To retrieve the _total_ volume instead in the ! present case, use the option 'VT': CALL TQGETR('VT ', 0, 0, RESULT, NOERR) WRITE(UNIT=*,FMT=11) 'Total V:', RESULT, ' dm^3'
Delta H: -0.25271E+06 J Delta V: 160.05 dm^3 Total V: 340.05 dm^3
! Compare the two values for the volume with the corresponding ones in ! the output table produced earlier. The volume printed at the top of ! such an output table is always the _total_ volume, whereas the line ! specifying the extensive property balance toward the end of the table ! shows the _change_ in volume. ! TQSTXP can be used to retrieve extensive property values for the ! incoming streams. Get the volume of the incoming CO2/CO stream: CALL TQSTXP('CO2CO ', 'V ', RESULT, NOERR) WRITE(UNIT=*,FMT=11) 'V(CO2CO): ', RESULT, ' dm^3' ! Get the value of the incoming air stream: CALL TQSTXP('O2N2 ', 'V ', RESULT, NOERR) WRITE(UNIT=*,FMT=11) 'V(O2N2): ', RESULT, ' dm^3'
V(CO2CO): 118.02 dm^3 V(O2N2): 61.974 dm^3
! For users of ChemApp versions prior to 3.2.0: Please note that the ! values returned by TQSTXP always er to the _last_ calculated ! equilibrium state. Even if one uses TQSTXP to retrieve properties of ! _incoming_ streams, one thus needs to perform an equilibrium ! calculation before the correct values are obtained. If TQSTXP is ! called before the first equilibrium calculation has been performed ! using TQCE or TQCEL, VAL will be zero. ! As of version 3.2.0 of ChemApp, TQSTXP can also be called _before_ the ! equilibrium calculation) ! As can be seen from the above results, this process, under the ! conditions we have chosen, results in solids to be formed in the ! reactor. We will now further investigate this systems by looking at ! how the amount of condensed material changes when ! a) the reaction temperature is varied, and ! b) the amount of air supplied is varied. ! **************************************************************** ! Varying the reaction temperature ! **************************************************************** ! Since several individual equilibria will be calculated, the results ! (temperature and amount of condensed phases) will be stored to a file ! for further processing. OPEN(11, FILE='caf103_1.rst', STATUS='unknown', IOSTAT=NOERR) IF (NOERR .NE. 0) THEN WRITE(*,*) 'ERROR: Cannot open result file.' STOP ENDIF ! Vary the temperature between 200 C and 1000 C in steps of 5 C DO J=200, 1000, 5 ! Use the variable TEMP to hold the current temperature. TEMP = J CALL TQSTEC('T ', 0, TEMP, NOERR) ! Calculate the equilibrium. In this case, we only need the value for the ! amount of condensed phases formed, theore TQCE is used and not ! TQCEL. CALL TQCE(' ', 0, 0, VALS, NOERR) ! Get the amount of condensed phases ACOND = GETACP(NPHASE) ! Write the temperature and the amount of condensed phases to the result ! file 12 FORMAT(2E12.5) WRITE(11,FMT=12) TEMP, ACOND ENDDO ! Close the result file CLOSE(11) ! When plotted, the resulting graph looks like the following Figure
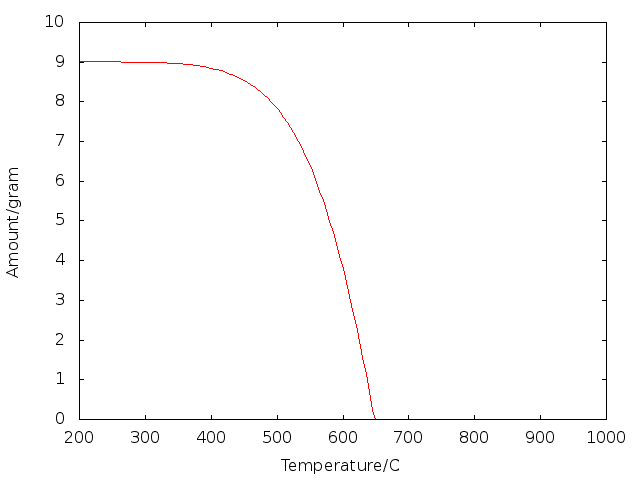
Calculated amount of condensed phases as a function of temperature. The amount of incoming air is kept constant. Note that the kink in the curve where it meets the zero amount axis would disappear if more individual points were calculated for the curve, or if the exact value of the temperature for a zero amount of condensed phases were explicitly determined using a target calculation.
! **************************************************************** ! Varying the amount of air supplied ! **************************************************************** ! Instead of the temperature, the amount of incoming air will be varied ! now. The temperature will be kept constant at the initial value of ! 600 C. To plot the results later on, we will again store them in a ! separate file. OPEN(11, FILE='caf103_2.rst', STATUS='unknown', IOSTAT=NOERR) IF (NOERR .NE. 0) THEN WRITE(*,*) 'ERROR: Cannot open result file.' STOP ENDIF ! Set the temperature for the equilibrium calculation (600 C) CALL TQSTEC('T ', 0, 600.D0, NOERR) ! Calculate 201 equilibria... DO J=0, 200 ! ... with the O2 amount varying between 0 and 1 mol... IAO2 = J * 0.005D0 ! ... and the N2 amount between 0 and 4 mol. IAN2 = IAO2 * 4.D0 ! To make sure ChemApp interprets these values as 'mol', set the amount ! unit accordingly CALL TQCSU('Amount ', 'mol ', NOERR) ! Set the amount of O2 in the air stream CALL TQSTCA('O2N2 ', 1, IO2, IAO2, NOERR) ! Set the amount of N2 in the air stream CALL TQSTCA('O2N2 ', 1, IN2, IAN2, NOERR) ! Calculate the equilibrium CALL TQCE(' ', 0, 0, VALS, NOERR) ! Set the amount unit to 'gram' CALL TQCSU('Amount ', 'gram ', NOERR) ! Get the amount of condensed phases ACOND = GETACP(NPHASE) ! Write the amounts of O2 and condensed phases to the result file WRITE(11,FMT=12) IAO2, ACOND ENDDO ! Close the result file CLOSE(11) ! When plotted, the resulting graph looks like the following Figure
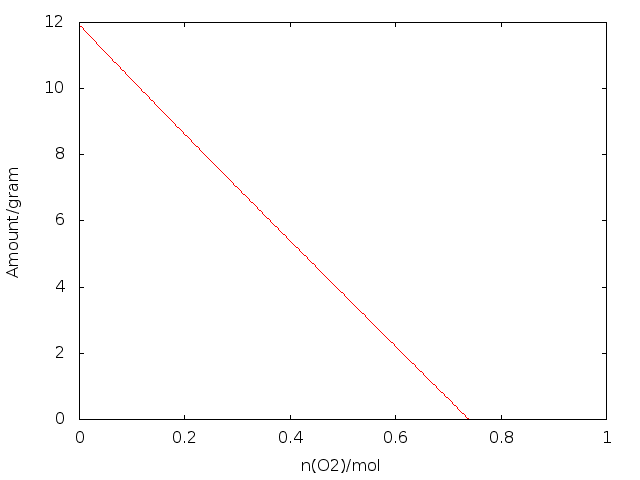
Calculated amount of condensed phases as a function of the O2 amount supplied. The temperature is kept constant at 600 C.
! **************************************************************** ! Varying the amount of air supplied _and_ the temperature ! **************************************************************** ! To investigate the combined influence of temperature and amount of air ! supplied on the resulting amount of condensed phases, both parameters ! are varied in nested loops. The results are again stored in a file for ! further processing. OPEN(11, FILE='caf103_3.rst', STATUS='unknown', IOSTAT=NOERR) IF (NOERR .NE. 0) THEN WRITE(*,*) 'ERROR: Cannot open result file.' STOP ENDIF ! The outer loop will vary the temperature between 200 C and 1000 C DO J=200, 1000, 20 TEMP = J CALL TQSTEC('T ', 0, TEMP, NOERR) ! The inner loop varies the amount of incoming air as above. DO K=0, 200, 5 IAO2 = K * 0.005D0 IAN2 = IAO2 * 4.D0 CALL TQCSU('Amount ', 'mol ', NOERR) ! Set the incoming amount of O2 CALL TQSTCA('O2N2 ', 1, IO2, IAO2, NOERR) ! Set the incoming amount of N2 CALL TQSTCA('O2N2 ', 1, IN2, IAN2, NOERR) CALL TQCE(' ', 0, 0, VALS, NOERR) CALL TQCSU('Amount ', 'gram ', NOERR) ! Get the amount of condensed phases ACOND = GETACP(NPHASE) ! Write the temperature and the amounts of O2 and condensed phases to ! the result file 13 FORMAT(3E12.5) WRITE(11,FMT=13) TEMP, IAO2, ACOND ENDDO WRITE(11,*) ENDDO ! Close the result file CLOSE(11) ! When plotted, the resulting graph looks like the following Figure
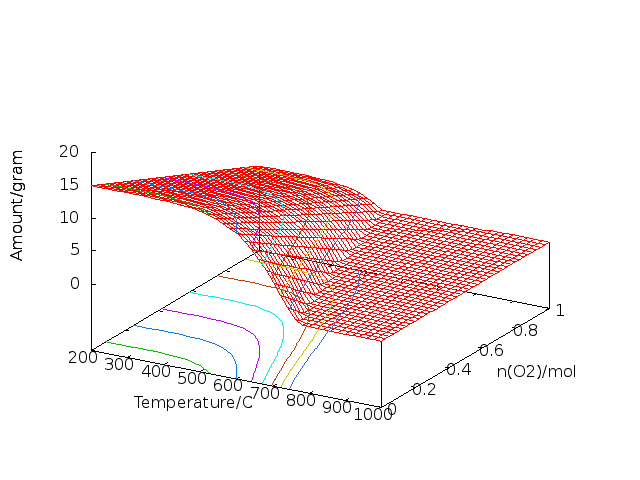
Calculated amount of condensed phases as a function of both the temperature and the O2 amount supplied. Interpolated contours for selected values of the amount of condensed phases have also been added by the plotting program. The contour for a zero amount of condensed phases (see Figure 9.4_5) is calculated explicitly below using target calculations.
! Check if we are working with the 'light' version. ! If we do, omit the following phase target calculation(s). CALL TQLITE(ISLITE, NOERR) IF (ISLITE .EQ. 1) THEN WRITE(*,FMT='(3(1X,A,/))') * '*** Phase target calculations have been omitted here,', * '*** since they are not possible with the', * '*** ''light'' version of ChemApp.' ELSE ! **************************************************************** ! Varying the amount of air supplied _and_ the temperature, searching ! for the explicit condition where the amount of condensed phase is zero. ! **************************************************************** ! Assuming that one is interested in the above process _not_ producing ! any condensed phases (i.e. complete combustion), it would be helpful ! to have a diagram which corresponds to the bottom plane of the above ! figure: A graph which, as a function of the incoming amount of air, ! shows the temperature at which the amount of condensed phases formed ! is zero. ! For the calculation procedure with ChemApp, this results in the ! following steps: ! 1) We are looking for the temperature at which condensed phases _just_ ! start to form. In terms of ChemApp, we thus have to define a ! precipitation target with respect to the gas phase. ! 2) For each value of the amount of air supplied, define the 'O2N2' ! stream with the proper amounts of O2 and N2. ! 3) ChemApp is then supposed to vary the temperature until the target ! (a phase precipitates from the gas phase) is met. Temperature is ! thus the target variable. This is specified as a parameter to TQCE, ! together with an estimated value. ! 4) The equilibrium is calculated for each of the amounts of air ! supplied, then the equilibrium temperature is retrieved and stored in ! the result file. OPEN(11, FILE='caf103_4.rst', STATUS='unknown', IOSTAT=NOERR) IF (NOERR .NE. 0) THEN WRITE(*,*) 'ERROR: Cannot open result file.' STOP ENDIF ! Reset the amount unit to 'mol' CALL TQCSU('Amount ', 'mol ', NOERR) ! Define the target as a precipitation target ('-1') for the gas ! phase (phase index 1) CALL TQSTEC('A ', 1, -1.D0, NOERR) ! The initial estimate for the temperature is passed via the first ! element of VALS to subsequent calls to TQCE VALS(1) = 600.D0 ! Vary the amount of supplied air DO J=0, 200 IAO2 = J * 0.005D0 IAN2 = IAO2 * 4.D0 ! Set the incoming amount of O2 CALL TQSTCA('O2N2 ', 1, IO2, IAO2, NOERR) ! Set the incoming amount of N2 CALL TQSTCA('O2N2 ', 1, IN2, IAN2, NOERR) ! Define the target variable (temperature), with the initial estimate in ! the first element of VALS, and calculate the equilibrium CALL TQCE('T ', 0, 0, VALS, NOERR) ! Retrieve the calculated equilibrium temperature CALL TQGETR('T ', 0, 0, TEMP, NOERR) ! Store the amount of O2 supplied and the calculated equilibrium ! temperature to the result file WRITE(11,FMT=12) IAO2, TEMP ENDDO ! Close data-file CLOSE(11) ! When plotted, the resulting graph looks like the following Figure ENDIF END
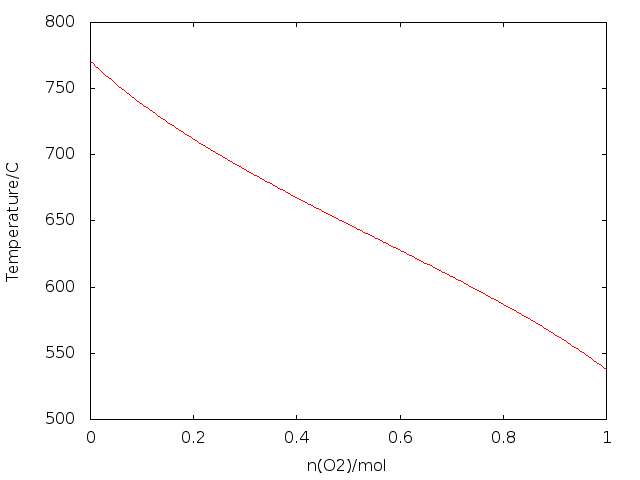
The curve represents the border between the area where condensed phases are formed (below the curve) and where the gas phase is the only one stable (above the curve). It represents the explicitly calculated contour for a zero amount of condensed phases in Figure 9.4_4.
Suggestions for enhancements
- Before adding to the code, answer the following questions for yourself:
- In the last part of the program, which looks at the conditions under which condensed phases are formed, why was a precipitation phase target used instead of a formation phase target?
- In the above example, the amount of air supplied was varied to investigate the influence of this parameter on the amount of condensed phases formed. Would it also be useful to vary the temperature of the air supplied and see what effect this has on the amount of precipitates formed?
- Assume that instead of preventing condensed phases to be formed, the minimisation of carbon monoxide (CO) formation is the primary concern. What influence do parameters like reaction temperature and air supply have in that case?
9.5 One-dimensional phase mapping
As of version 3.0.0, ChemApp is able to perform one-dimensional phase mapping calculations. Users of FactSage will very likely be familiar with this feature already, since it is very useful in phase equilibrium calculations. One-dimensional phase mapping is generally used to search for phase boundaries within the range of a given variable (temperature, pressure, or composition). With only a few lines of code it allows the programmer to automatically locate every phase boundary within a specified range.
Using one-dimensional phase mapping thus makes it very easy to find phase boundaries in a variety of phase diagrams, such as those with temperature/composition (T/x), pressure/composition (P/x), and temperature/pressure (T/P) as axes.
The following program demonstrates one-dimensional phase mapping calculations in a T/x and a T/P diagram.
The system Pb-Sn, already described in the worked example Phase equilibria and phase target calculations, is used to demonstrate the calculation of two one-dimensional phase maps in a T/x diagram, with the temperature as the search variable. The lines marked 1 and 2 in Figure 9.5_1 indicate the range within which the phase boundaries will be searched.
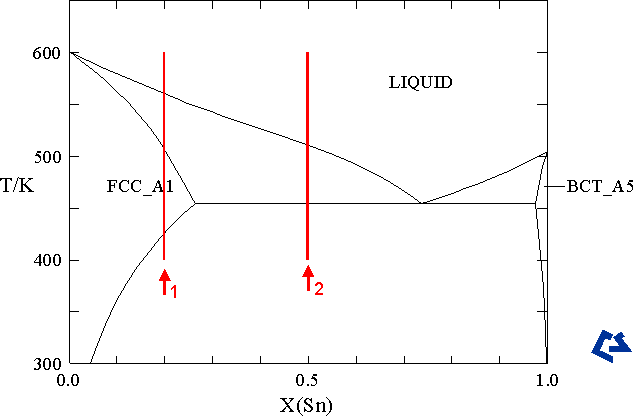
The Pb-Sn phase diagram
For the system System Fe2SiO4-Mg2SiO4 (see FEMGSIO4.DAT (System Fe2SiO4-Mg2SiO4)), a T/P phase diagram has been calculated, of which a section is shown in Figure 9.5_2. In the course of the following example program, two one-dimensional phase maps will be calculated in this phase diagram, using first pressure (line 1) and then composition (line 2) as the search variable.
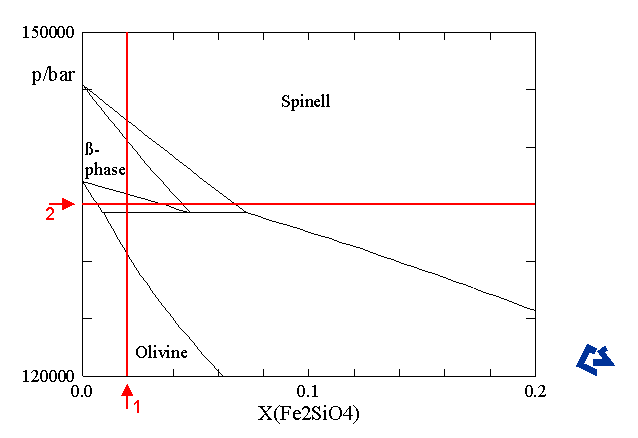
The Fe2SiO4-Mg2SiO4 pressure/composition phase diagram at 1000 K
Definition of goals
In this worked example emphasis is placed on demonstrating the method of finding the phase boundaries within the ranges of the search variables specified. For all the phase boundaries found, only minimal tabular information is output, since the retrieval of results from a phase equilibrium calculation has been demonstrated elsewhere (see for instance TQGETR). For the first phase map it is also demonstrated which results need to be collected to produce a graphical representation of the phase map.
The following one-dimensional phase maps will be calculated:
- In the T/x phase diagram Pb-Sn, two maps with temperature as the search variable (lines 1 and 2 in Figure 9.5_1). The first one of these two will be shown graphically.
- In the T/P phase diagram Fe2SiO4-Mg2SiO4, a map with pressure as the search variable (line 1 in Figure 9.5_2)
- For the same system and phase diagram, a map with the concentration as search variable (line 2 in Figure 9.5_2)
Tools and concepts used
For the present program example, only one ChemApp subroutine is introduced that has not been used in previous worked examples: TQMAP. Since performing one-dimensional phase mapping calculations is in some ways similar to phase target calculation, the options available for calls to TQMAP are similar to those of TQCE. They differ in one important detail though: Whereas a phase target calculation, performed with TQCE, results in a singular result (normally the equilibrium at the phase boundary found), a one-dimensional phase mapping calculation consists of multiple results, where each call to TQMAP retrieves one of them.
For the programmer this means that after every call to TQMAP, a subroutine parameter (named ICONT in the example below) is returned which indicates whether there are more phase boundaries to come. Thus TQMAP is usually called in a loop, which is left when ICONT indicates that there are no more phase boundaries left.
Implementation
! One-dimensional phase mapping calculations ! Note that this program contains phase target calculations, ! which cannot be performed with the 'light' version of ChemApp ! Subroutine BSORT implements a simple bubble sort algorithm. It is ! used to sort the array of temperature values for which phase ! equilibria are to be calculated. SUBROUTINE BSORT(NPTS, TARRAY, NUSED) IMPLICIT NONE INTEGER NPTS, NUSED DOUBLE PRECISION TARRAY(NPTS) INTEGER I,J DOUBLE PRECISION TEMP ! Sort the first NUSED elements of TARRAY, which has a total number ! of NPTS elements, in ascending order DO I=1,NUSED DO J=I+1, NUSED IF(TARRAY(I).GT.TARRAY(J)) THEN TEMP=TARRAY(I) TARRAY(I)=TARRAY(J) TARRAY(J)=TEMP ENDIF ENDDO ENDDO RETURN END ! Subroutine PSP prints the value of the 'search variable' (temperature, ! pressure, or composition), the names of the stable phases, and their ! amounts. It is used after every call to TQMAP. SUBROUTINE PSP(VALUE1, VALUE2) IMPLICIT NONE DOUBLE PRECISION VALUE1, VALUE2, AMT, ACT, EPS CHARACTER PNAME*24, PSTAT*24 INTEGER NPHASE, I, NOERR PARAMETER(EPS = 1.E-8) ! If the second parameter to PSP is zero, we only need to print the ! first one. IF (VALUE2 .NE. 0.D0) THEN WRITE(6,FMT='(2F12.3)') VALUE1, VALUE2 ELSE WRITE(6,FMT='(F12.2)') VALUE1 ENDIF ! Find out how many phases we have in the system CALL TQNOP(NPHASE, NOERR) ! For every phase, check whether its activity is unity. Note that for ! numerical reasons, ChemApp will sometimes return an activity of less ! than unity for a stable phase. To still be able to determine whether a ! phase is stable, one should be on the safe side if the phase's ! activity is compared against 1.0 - 1E-8, as in the IF-clause below. If ! the activity is larger than this value, the phase can be considered to ! be stable. DO I=1, NPHASE CALL TQGETR('AC ', I, 0, ACT, NOERR) IF (ACT .GE. (1.D0 - EPS)) THEN ! Check if the phase has the status ENTERED, otherwise (i.e. when the ! status of the phase is DORMANT) it might have an activity > 1 and ! still have an equilibrium amount of zero: CALL TQGSP(I, PSTAT, NOERR) IF (PSTAT .EQ. 'ENTERED') THEN ! If it is stable _and_ its status is ENTERED, get the name of that ! phase... CALL TQGNP(I, PNAME, NOERR) ! ...and its equilibrium amount... CALL TQGETR('A ', I, 0, AMT, NOERR) ! ... and print both: WRITE(*,FMT='(27X,A,F8.2)') PNAME, AMT ENDIF ENDIF ENDDO RETURN END PROGRAM CAF104 IMPLICIT NONE INTEGER NOERR, NUMCON, BCT2, IPB, ISN, IPFESI, IPMGSI, ISLITE, * ICONT, RESNO, NUSED, ASIZE, ITEMP PARAMETER(ASIZE = 100) DOUBLE PRECISION VALUE1, VALUE2, VALS(2), TARRAY(ASIZE), * EPS, AMTS(4), ACTS(4) PARAMETER(EPS = 1.E-3) NUSED = 0 ! Initialise ChemApp CALL TQINI(NOERR) ! Open the thermochemical data-file pbsn.dat (system Pb-Sn) ! for reading CALL TQOPNA('pbsn.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Check if we are working with the 'light' version. ! If we do, omit the following phase target calculation(s). CALL TQLITE(ISLITE, NOERR) IF (ISLITE .EQ. 1) THEN WRITE(*,FMT='(3(1X,A,/))') * '*** Phase target calculations have been omitted here,', * '*** since they are not possible with the', * '*** ''light'' version of ChemApp.' ELSE ! The data-file for system Pb-Sn contains a second copy of the BCT_A5 ! phase, because of the inherent metastable miscibility gap. Since this ! miscibility gap will stay metastable under the conditions used in this ! example, we will eliminate the second copy of that phase, which means ! it will be disregarded completely in the following calculations. CALL TQINP('BCT_A5#2', BCT2, NOERR) CALL TQCSP(BCT2, 'ELIMINATED ', NOERR) ! Determine the index numbers for the system components Pb and Sn CALL TQINSC('Pb ', IPB, NOERR) CALL TQINSC('Sn ', ISN, NOERR) ! As indicated in Figure above (The Pb-Sn phase diagram), label "1", the first phase map ! should be done with the temperature as the search variable, for ! X(Sn)=0.2. ! Set the incoming amounts for the Sn and Pb CALL TQSETC('IA ', 0, ISN, 0.2D0, NUMCON, NOERR) CALL TQSETC('IA ', 0, IPB, 0.8D0, NUMCON, NOERR) ! VALS is passed to TQMAP; this array of 2 DOUBLE PRECISION variables ! contains the lower and upper values of the search variable. Note that ! the first two calls to TQMAP _always_ calculate the equilibria at ! VALS(1) and VALS(2), and thus do _not_ constitute phase boundaries. VALS(1) = 400.D0 VALS(2) = 600.D0 ! The purpose of the array TARRAY is to hold the temperature values at ! which the equilibria will be calculated later. This set of temperature ! values consists of ! 1) a number of evenly spaced temperature intervals, ! 2) and the temperatures at which TQMAP finds phase boundaries. ! Part of the array is first filled with evenly spaced temperature ! intervals. As upper and lower values we use the same values as we did ! for the initialisation of VALS(1) and VALS(2) above. The variable ! NUSED keeps track of how many items of the array are actually used: DO ITEMP = 400, 600, 10 NUSED = NUSED + 1 TARRAY(NUSED) = ITEMP ENDDO ! RESNO keeps track of the number of results we get from TQMAP. This ! allows us to ignore the first two temperatures TQMAP reports. As ! indicated above, the first two calls to TQMAP do _not_ give us ! information about a phase boundary, but on the state at the lower and ! upper end of the search interval (i.e. at VALS(1) and VALS(2)). Since ! we have these temperatures in our temperature array TARRAY already ! (see the DO loop above), we only need to pay attention when we ! retrieve the third and any subsequent result with TQMAP. RESNO = 0 ! First call to TQMAP. Note the option 'TF ', where 'T' indicates that ! temperature is the search variable, and 'F' ('first') that it is the ! first call to TQMAP for the current phase map. The variable returned ! through ICONT with every call to TQMAP will allow us to decide whether ! more calls to TQMAP are necessary. Since this is only the first call ! to TQMAP, we know anyway that we have to continue. CALL TQMAP('TF ',0,0,VALS,ICONT,NOERR) RESNO = RESNO + 1 ! Get the temperature. Since it is the first call to TQMAP, we _know_ ! that this will be the value of VALS(1) CALL TQGETR('T ',0,0,VALUE1,NOERR) 100 FORMAT(1X,A,T28,A,T50,A) ! Print a header for the table to come. WRITE(*,FMT=100) 'Temperature/K', 'Stable phases', 'Amount/mol' ! Call PSP (print stable phases), passing the value for the temperature ! we just retrieved. PSP will print a formatted entry for the result ! table. CALL PSP(VALUE1, 0.D0) ! TQMAP is called again. Note the option 'TN ', where 'T' indicates that ! temperature is the search variable, and 'N' ('next') that it is _not_ ! the first call to TQMAP for the current phase map. 20 CALL TQMAP('TN ',0,0,VALS,ICONT,NOERR) RESNO = RESNO + 1 ! Get the temperature. CALL TQGETR('T ',0,0,VALUE1,NOERR) ! If this is the second call to TQMAP, we know that the temperature ! retrieved here is VALS(2), and also that TARRAY already contains ! it. If, on the other hand, we are already retrieving our third or any ! subsequent result (checked through RESNO), we know it is a phase ! boundary. IF (RESNO .GT. 2) THEN ! What follows here is necessary because we would like to see the ! results of this one-dimensional phase map in a graph later. ! If we were just interested in the _position_ of this phase boundary, ! and thus the _value_ of the search variable (the temperature in this ! case), it would be enough to record this piece of information. ! However, since we would like to see a graphical representation of the ! phase map, we might not get the desired results if we just add the ! temperature found to TARRAY. Instead of calculating the equilibrium at ! the temperature just retrieved, we better calculate it at _two_ ! temperatures: One slightly below, and one slightly above the ! temperature found. Thus these two temperatures are added to TARRAY ! instead of the exact temperature for the phase boundary (EPS has been ! defined using a PARAMETER statement at the beginning of the program): NUSED = NUSED + 1 TARRAY(NUSED) = VALUE1 - EPS NUSED = NUSED + 1 TARRAY(NUSED) = VALUE1 + EPS ENDIF ! Print the entry for the result table. For this purpose, we need the ! _exact_ temperature at the phase boundary, as determined by TQMAP: CALL PSP(VALUE1, 0.D0) ! The variable ICONT returned by TQMAP indicates whether we need to make ! any more calls to TQMAP, i.e. whether there are any more phase ! boundaries within the search interval we specified. If ICONT is ! positive, we need to continue. IF (ICONT .GT. 0) GOTO 20
Temperature/K Stable phases Amount/mol
400.00
BCT_A5#1 0.06
FCC_A1 0.94
600.00
LIQUID 1.00
426.36
BCT_A5#1 0.00
FCC_A1 1.00
505.37
LIQUID 0.00
FCC_A1 1.00
560.01
LIQUID 1.00
FCC_A1 0.00
! When we arrive at this point of the program ICONT is zero, which tells ! us that there are no more phase boundaries left within the search ! interval we specified. ! The array TARRAY now contains all the temperatures at which we want to ! calculate the equilibria for our graphical phase map: the evenly ! spaced temperatures plus the two temperatures for every phase ! boundary: One slightly below and one slightly above the temperature of ! the original phase boundary. ! ! The only problem is that these temperatures are not in ascending ! order, since all the temperatures for the phase boundaries have simply ! been added to the end of the list of evenly spaced temperatures. ! ! This is revealed by looking at the first and last 10 values of TARRAY: 101 FORMAT('TARRAY(',I2,') = ',F12.5) DO ITEMP=1, NUSED IF ((ITEMP .LT. 10) .OR. (ITEMP .GT. (NUSED-10))) THEN WRITE(*,101) ITEMP, TARRAY(ITEMP) ENDIF ENDDO
TARRAY( 1) = 400.00000 TARRAY( 2) = 410.00000 TARRAY( 3) = 420.00000 TARRAY( 4) = 430.00000 TARRAY( 5) = 440.00000 TARRAY( 6) = 450.00000 TARRAY( 7) = 460.00000 TARRAY( 8) = 470.00000 TARRAY( 9) = 480.00000 TARRAY(18) = 570.00000 TARRAY(19) = 580.00000 TARRAY(20) = 590.00000 TARRAY(21) = 600.00000 TARRAY(22) = 426.35862 TARRAY(23) = 426.36062 TARRAY(24) = 505.36777 TARRAY(25) = 505.36977 TARRAY(26) = 560.00807 TARRAY(27) = 560.01007
! To have the values for the graph displayed properly later, we thus ! need to sort this array using the subroutine BSORT: CALL BSORT(ASIZE, TARRAY, NUSED) ! We are now ready to calculate the equilibria for all the temperatures ! in TARRAY. These equilibrium calculations now being performed are ! _not_ target calculations of any kind any more, that's why we reset ! _all_ previous conditions including all targets and target variables. CALL TQREMC(-2, NOERR) ! The following equilibria are to be calculated for the same alloy ! concentration of course, so we use the same incoming amounts again. CALL TQSETC('IA ', 0, ISN, 0.2D0, NUMCON, NOERR) CALL TQSETC('IA ', 0, IPB, 0.8D0, NUMCON, NOERR)
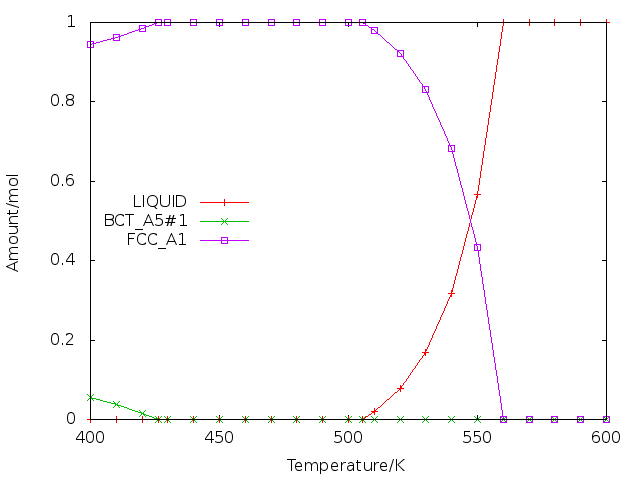
One-dimensional phase mapping calculation in the system Pb-Sn at xSn = 0.2: Calculated phase amounts as a function of temperature. Although the x-resolution is rather low (note the slight kinks in the curve between 510 K and 560 K) the temperatures at which phases appear or disappear, i.e. at which the curves reach unity or zero, have been considered specifically.
! The graphical representation of the one-dimensional phase map will be ! done using two x/y graphs: one displaying the phase amounts vs. the ! temperature, the other one the phase activities vs. the ! temperature. We will write two output data-files for this purpose: ! caf104_1.rst will contain the phase amounts, whereas caf104_2.rst will ! contain the phase activities. OPEN(11, FILE='caf104_1.rst', STATUS='unknown', IOSTAT=NOERR) IF (NOERR .NE. 0) THEN WRITE(*,*) 'ERROR: Cannot open result file.' STOP ENDIF OPEN(12, FILE='caf104_2.rst', STATUS='unknown', IOSTAT=NOERR) IF (NOERR .NE. 0) THEN WRITE(*,*) 'ERROR: Cannot open result file.' STOP ENDIF ! For every temperature in TARRAY, calculate the equilibrium: DO ITEMP=1, NUSED CALL TQSETC('T ', 0, 0, TARRAY(ITEMP), NUMCON, NOERR) ! Note again that these are regular equilibrium calculations, no targets ! are involved: CALL TQCE(' ', 0, 0, VALS, NOERR) ! Retrieve the equilibrium amounts of _all_ phases with a single call to ! TQGETR. Note that AMTS is an array of appropriate size. CALL TQGETR('A ', -1, 0, AMTS, NOERR) ! Write the temperature plus the phase amounts to the first output ! data-file: WRITE(11,FMT='(5F15.10)') TARRAY(ITEMP), AMTS ! Retrieve the equilibrium activities of _all_ phases with a single call ! to TQGETR. Note that ACTS is also an array of appropriate size. CALL TQGETR('AC ', -1, 0, ACTS, NOERR) ! Write the temperature plus the phase activities to the second output ! data-file: WRITE(12,FMT='(5F15.10)') TARRAY(ITEMP), ACTS ENDDO ! We have now calculated the equilibria for all temperature values in ! TARRAY and can close the two output data-files. CLOSE(11) CLOSE(12)
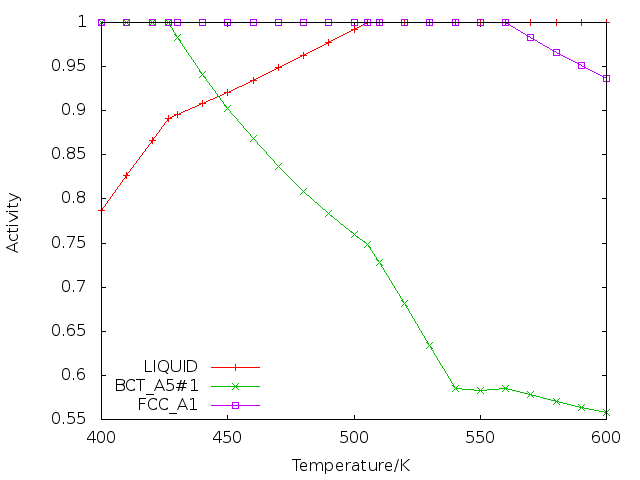
One-dimensional phase mapping calculation in the system Pb-Sn at xSn = 0.5: Calculated phase activities as a function of temperature. The kink in the activity curve for BCT_A5#1 near 540 K is due to the metastable miscibility gap in that phase.
! When plotted, the two graphs, displaying the equilibrium phase amounts ! and phase activities vs. temperature look like Figures 3 and 4 above. ! For the next one-dimensional phase mapping calculations we will only ! use tabular output. First, remove all previously set conditions. CALL TQREMC(-2, NOERR) ! We will now calculate another one-dimensional phase map in the same ! system, again with temperature as the search variable. The new ! alloy composition will cause TQMAP to find the eutectic. CALL TQSETC('IA ', 0, ISN, 0.5D0, NUMCON, NOERR) CALL TQSETC('IA ', 0, IPB, 0.5D0, NUMCON, NOERR) ! The temperature search interval used is the same: VALS(1) = 400.D0 VALS(2) = 600.D0 ! First call to TQMAP (note again the 'F' in the option parameter) CALL TQMAP('TF ',0,0,VALS,ICONT,NOERR) ! Retrieve the temperature, which we already know is VALS(1) CALL TQGETR('T ',0,0,VALUE1,NOERR) ! Print a header for the table to come. WRITE(*,FMT=100) 'Temperature/K', 'Stable phases', 'Amount/mol' ! Call PSP to print the information on the stable phases: CALL PSP(VALUE1, 0.D0) ! TQMAP is called with 'N' in the option parameter: 30 CALL TQMAP('TN ',0,0,VALS,ICONT,NOERR) ! Get the temperature... CALL TQGETR('T ',0,0,VALUE1,NOERR) ! ... and print the entry for the result table: CALL PSP(VALUE1, 0.D0) IF (ICONT .GT. 0) GOTO 30
Temperature/K Stable phases Amount/mol
400.00
BCT_A5#1 0.42
FCC_A1 0.58
600.00
LIQUID 1.00
454.56
LIQUID 0.00
BCT_A5#1 0.33
FCC_A1 0.67
510.45
LIQUID 1.00
FCC_A1 0.00
! Note that the above table lists a 3-phase equilibria now, which for the ! current system means we found the eutectic temperature (compare this ! with the results for the same equilibrium determined in worked example ! 3: Phase equilibria and phase target calculations). ! So far we have calculated one-dimensional phase maps for a ! temperature/composition phase diagram, in our case keeping the ! composition (and implicitly the pressure) constant and using the ! temperature as the search variable. The next phase map we will ! calculate at a constant temperature, this time varying the ! pressure. For this purpose, we will use a different thermochemical ! data-file for the system Fe2SiO4-Mg2SiO4 which includes ! pressure-dependent Gibbs-energy data. ! Open the thermochemical data-file femgsio4.dat (system Fe2SiO4-Mg2SiO4) ! for reading CALL TQOPNA('femgsio4.dat', 10, NOERR) ! Read data-file CALL TQRFIL(NOERR) ! Close data-file CALL TQCLOS(10, NOERR) ! Determine the index numbers for the stoichiometric compounds Fe2SiO4 ! and Mg2SiO4. These two phases will be used to define the incoming ! amounts for the next calculations. Note that they are included in the ! thermochemical data-file only for this purpose (to allow for a ! somewhat easier definition of incoming amounts), since the Gibbs ! energy functions for these two stoichiometric compounds are only ! dummies. CALL TQINP('Fe2SiO4 ', IPFESI, NOERR) CALL TQINP('Mg2SiO4 ', IPMGSI, NOERR) ! Select a composition of 2 mol-% Fe2SiO4, the rest is Mg2SiO4: CALL TQSETC('IA ', IPFESI, 0, 0.02D0, NUMCON, NOERR) CALL TQSETC('IA ', IPMGSI, 0, 0.98D0, NUMCON, NOERR) ! Select the range for the search variable, which is the pressure in the ! current case. We will search for any phase boundaries between 120 kbar ! and 150 kbar (see also the line marked '1' in Figure 2 above. vals(1) = 120000.D0 vals(2) = 150000.D0 ! The temperature is set to 1000 K CALL TQSETC('T ', 0, 0, 1000.D0, NUMCON, NOERR) ! First call to TQMAP. Note the option 'PF ', where 'P' indicates that ! pressure is the search variable, and 'F' that it is the first call ! to TQMAP for the current phase map. CALL TQMAP('PF ', 0, 0, VALS,ICONT, NOERR) ! Get the pressure. Since it is the first call to TQMAP, we _know_ ! that this will be the value of VALS(1) CALL TQGETR('P ',0,0,VALUE1,NOERR) ! Print a header for the table to come. WRITE(*,FMT=100) 'Pressure/bar', 'Stable phases', 'Amount/mol' ! Call PSP (print stable phases), passing the value for the pressure ! we just retrieved. PSP will print a formatted entry for the result ! table. CALL PSP(VALUE1, 0.D0) ! TQMAP is called again. Note the option 'PN ', where 'P' indicates that ! temperature is the search variable, and 'N' that it is _not_ the first ! call to TQMAP for the current phase map. 40 CALL TQMAP('PN ',0,0,VALS,ICONT,NOERR) ! Get the pressure. CALL TQGETR('P ',0,0,VALUE1,NOERR) ! Print the entry for the result table. CALL PSP(VALUE1, 0.D0) ! If the value of ICONT is positive, we need to call TQMAP again... IF (ICONT .GT. 0) GOTO 40
Pressure/bar Stable phases Amount/mol
120000.00
Olivine 2.00
150000.00
Spinell 2.00
130551.81
Olivine 2.00
Spinell 0.00
134268.49
Olivine 1.66
Spinell 0.34
beta-phase 0.00
135878.02
Olivine 0.00
beta-phase 2.00
140455.56
Spinell 0.00
beta-phase 2.00
142313.61
Spinell 2.00
beta-phase 0.00
! The result table is complete now. Compare the phase boundaries found ! with the phase diagram in Figure 2 above. Note that the total ! amount in the system is always calculated to be 2 mol. This is due to ! the fact that the phase constituents of all mixture phases in this ! data-file (Spinell, Olivine, and \DF-phase) have been scaled with the ! factor 0.5 in order to obtain the correct ideal entropy between Fe and ! Mg. ! The last one-dimensional phase map will use composition as the search ! variable, keeping both temperature and pressure constant. As with ! other phase target calculations which use composition as the target ! variable, the input in this case is a bit more complicated (see also ! worked example 3: Phase equilibria and phase target calculations). ! This is due to the fact that when one is interested in comparing the ! result for this type of calculation with graphical phase diagram ! information, it is necessary to keep the total substance amount in the ! system constant (i.e. use concentrations rather than absolute ! amounts). ! First, reset all conditions and targets again: CALL TQREMC(-1,NOERR) ! Set the pressure to 135 Kbar (see Figure 2 above): CALL TQSTEC('P ',0,135000.D0,NOERR) ! As explained above, we need to make more than one call to TQMAP to ! tell ChemApp about our choice of the search variable ranges. In the ! present case, we are working with a (pseudo-)binary system, so no more ! than two calls are necessary. ! ! With the first call, we define the composition range for Fe2SiO4 as ! having a minimum of 0 mol and a maximum of 0.2 mol, compare again to ! Figure 2 above. The option 'IA0 ' is the same one as used in ! phase target calculations with composition as target variable (see ! again worked example 3: Phase equilibria and phase target ! calculations), and makes sure that TQMAP does _not_ actually calculate ! anything at this point ('0'), but only accepts the values passed as ! information on the incoming amounts ('IA'). VALS(1) = 0.0D0 VALS(2) = 0.2D0 CALL TQMAP('IA0 ',IPFESI,0,VALS,ICONT,NOERR) ! Now we need to tell ChemApp to vary the composition of Mg2SiO4 ! accordingly, keeping the total substance amount in the system ! constant. For the range of Fe2SiO4 selected above, this means setting ! VALS(1) to 1 mol and VALS(2) to 0.8 mol: VALS(1) = 1.0D0 VALS(2) = 0.8D0 ! In the subsequent call to TQMAP, the option 'IAF ' is used, meaning ! that again information on incoming amounts is passed ('IA'), but this ! time ChemApp is supposed to start calculation and determine the first ! result ('F'): CALL TQMAP('IAF ',IPMGSI,0,VALS,ICONT,NOERR) ! Retrieve the composition for the first result. CALL TQGETR('IA ',IPFESI,0,VALUE1,NOERR) CALL TQGETR('IA ',IPMGSI,0,VALUE2,NOERR) ! Print a header for the table to come. WRITE(*,FMT=100) ' X(Fe2SiO4) X(Mg2SiO4)', 'Stable phases', * 'Amount/mol' ! Pass both concentrations to PSP to print the table entry. CALL PSP(VALUE1, VALUE2) ! TQMAP is called again. Note the option 'IAN ', where 'IA' indicates ! that composition is the search variable, and 'N' that it is _not_ the ! first call to TQMAP for the current phase map. 50 CALL TQMAP('IAN ',0,0,VALS,ICONT,NOERR) ! Retrieve the composition... CALL TQGETR('IA ',IPFESI,0,VALUE1,NOERR) CALL TQGETR('IA ',IPMGSI,0,VALUE2,NOERR) ! ... and add an entry to the output table. CALL PSP(VALUE1, VALUE2) ! If the value of ICONT is positive, we need to call TQMAP again... IF (ICONT .GT. 0) GOTO 50
X(Fe2SiO4) X(Mg2SiO4) Stable phases Amount/mol
0.000 1.000
Olivine 2.00
0.200 0.800
Spinell 2.00
0.007 0.993
Olivine 2.00
beta-phase 0.00
0.035 0.965
Olivine 0.00
beta-phase 2.00
0.044 0.956
Spinell 0.00
beta-phase 2.00
0.068 0.932
Spinell 2.00
beta-phase 0.00
! When ICONT does not have a positive value any more, we know that we ! have found all possible phase boundaries within the search interval ! specified. ENDIF END
Suggestions for enhancements
- Before adding to the code, answer the following questions for yourself:
- As mentioned already, the results obtained from the first two calls of TQMAP for every one-dimensional phase map do not represent information about a phase boundary found. Looking at any of the tabular outputs in the above example, why is that obvious, even without knowing about this behaviour explicitly beforehand, and assuming one does not happen to recognise the values for VALS(1) and VALS(2)?
- In preparation for the calculations which lead to the graphical representation of the one-dimensional phase map for the system Pb-Sn, the determined temperatures of the various phase boundaries were not added to TARRAY. Instead, two values were added: one slightly below, and one slightly above the temperature found. Why is that not really necessary in the present case? For which kind of phase maps does it make a notable difference though when graphs are produced? Hint: look at the example code in the manual section for TQMAP.
- Produce graphical output for the other three calculated one-dimensional phase maps.
- In the above code, substitute calls to TQMAP with TQMAPL and compare the type of ChemSage/ChemApp output tables produced with those resulting from phase target calculations using TQCEL.
10 Thermodynamic Data - Sources and Quality Considerations
In order to perform calculations using ChemApp, apart from the ChemApp object code you also need thermochemical data. These data are required in the form of thermochemical data-files in ChemApp-compatible format. The sample data-file cosi.dat supplied with ChemApp is used for the instruction contained in this part of the manual. Please contact GTT-Technologies if you need more information on thermochemical data, especially on non-ideal solution models.
10.1 Sources of thermochemical data
There are several sources of thermochemical data:
- Supplied as databases for use with FactSage (see Table Thermochemical data available from GTT-Technologies). If you are a user of FactSage, you can prepare an appropriate data-file for ChemApp from the databases.
- As extracts in the form of ready-to-use data-files available from GTT-Technologies (see Table Thermochemical data available from GTT-Technologies).
- Use your own data to compile a data-file in the required format, if you are not a FactSage user and wish to prepare your own data. However, note that you need to be well familiar with the structure and contents of such data-files. Please contact GTT-Technologies before you start preparing a file. GTT also offers a program called CSFAP that provides an easy-to use graphical interface to edit, enhance, and merge data-files in ASCII format.
| Major databases |
| SGTE pure substances database |
| SGTE solution database |
| FACT pure substances database |
| FACT solution database |
| Specialty databases |
| SGTE noble metals database |
| SGTE solders database |
| SGTE nuclear database |
| FACT light metal intermetallic compounds |
| FactSage steel database |
| FactSage Cu-rich alloy database |
| FactSage lead database |
| FactSage ultra-pure silicon database |
| Thermodata nuclear database |
| Data-files |
| 'Off-the-shelf' data-files for direct use |
| Customised data-files compiled to user specification, for direct use, with data from the above databases and other sources |
10.2 Quality considerations
It is very important when applying thermodynamic data, especially for critical applications, that the user understands that certain limitations may exist regarding its quality and the generality of its use; this applies especially to complex data-files for multicomponent, multiphase systems. These potential limitations apply whether data is supplied by GTT, or whether you prepare it yourself. It is important that all data be verified against known facts, wherever possible.
Quality categories
The most comprehensive (and thus reliable) kind of data-file to use is one that is fully consistent; i.e. it contains the Gibbs energies of all phases of the chemical system given as functions of temperature, pressure, and composition based on the standard element reference state (SER). Such a data-file permits calculation of phase equilibria for all temperatures and pressures from room temperature upwards and over the entire composition range covered by the elementary components; also, the thermodynamic properties of any of the phases in the system - integral or partial values of specific heat, enthalpy, entropy, Gibbs energy or volume. However, such a data-file is an ideal case. The higher the number of components in a system the more likely it is that certain simplifications are necessary.
These simplifications relate to restrictions of the range of applicability, i.e. temperature and composition, and also to the range of thermodynamic properties that can be calculated. These restrictions are perfectly permissible when they match the scope of the application. The following examples demonstrate this point further:
- Calculation of equilibria for highly oxidising conditions. In this case, it is not necessary that metallic subsystems be considered in the calculation and data for them need not be included in the data-file. Hence, it would not present a problem if these data were not readily available.
- Calculation of phase equilibria only. It is possible to use a data-file based only on relative Gibbs energies with a simple A+B*T temperature relationship.
- A calculation involves a multi-component system with one major component. The phases for this component can be described with a dilute solution approach. This restricts the composition range covered by the data-file considerably, but in most cases reliable information can be gained within the permitted range.
10.3 Techniques for compiling a data-file
Preparation of any data-file implies that at least some, if not all, thermodynamic data are available in some form, preferably fully assessed and qualitatively approved. In these cases, compilation of a high quality, fully consistent data-file is relatively straightforward. However, it is frequently the case that these kind of data are not immediately at hand. In such cases, recourse has to be made to other approaches:
- to carry out a search in the technical literature for experimental information. This enables an assessment to be made and a data-file can be prepared that will meet one of the above criteria.
- to use theoretical techniques in order to estimate the required data. In this case, the reliability of estimated data is not comparable with that based on a full assessment using comprehensive experimental data.
As may be understood from the above, preparation of a thermochemical data-file requires a high level of knowledge, skill and experience. Even when these attributes are present, it is not always possible to produce a data-file that immediately completely matches all a user's requirements.
The problems associated with the reliability of a data-file assume another level of significance when a person inexperienced in the field attempts to compile a new data-file from available thermochemical data, or even to compile a data-file for a more comprehensive system from two or more smaller data-files. A number of factors can be of major importance; for example, the presence of miscibility gaps and the relative importance of missing data for phases, etc. Hence, GTT-Technologies offers users a comprehensive data service, either to provide data-files of acceptable quality , or to provide assistance in their compilation and checking.
10.4 GTT data-file identification
If you receive data-files from GTT-Technologies, apart from the example data-files distributed with ChemApp, you will note that they are identified with a coded serial number made up of letters and digits; for example, 0135B03T.DAT. The code provides you with the following information:
- a four digit designation number, unique to the system comprising the data-file
- a quality classification code (letter)
- a single digit version number (0 for the first release; 1, 2,... for upgrades)
- a single digit for the year of generation or upgrading (5 for 1995, etc.)
- a single letter code for the originator (G for GTT, T for the Institute for Theoretical Metallurgy, RWTH Aachen, S for SGTE, etc.)
- a standard extension name .DAT applies to all data-files.
Hence, from the serial number, it is possible for you to know the quality of a data-file, if and when it was upgraded, and the originator of the original or upgraded data-file. The code used to designate the quality of GTT data-files is shown in Table below.
| Quality | Definition |
|---|---|
| A | fully consistent thermodynamic data covering the entire range of temperature and composition |
| B | partially consistent thermodynamic data covering only a specified range of temperature and/or composition of the system, a very high proportion of the data is reliably assessed |
| C | partially consistent data covering only a specific range of temperature and/or composition, a significant amount of estimated data is used |
| Z | data-files under development - a reliable quality definition is not yet possible |
Example of a data-file serial number
0135B03T.DAT signifies that the original version of the data-file 0135 was produced in 1993, has the quality designation 'B', and was produced by co-workers in the (T)HERDAS group at the Institute for Theoretical Metallurgy at the RWTH Aachen.
0135A24G.DAT means that the same data-file as in the previous example has been upgraded by (G)TT to 'A' quality after a second revision, the last one being in 1994.
Documentation of data-files
In addition to the serial number, each GTT data-file is comprehensively documented. A commentary section below the thermochemical data provides details of the history, quality, area(s) of application, sources of data, literature references, details of the upgrading of the data-file, etc. In this way, a complete documentation of the data-file is provided for the user to judge the suitability of a data-file for particular applications. If the data-file is supplied in "transparent format" (see Transparent data-files), a separate file with the comments is provided.
11 Thermochemical Data-files - Structure and Handling
Thermochemical data-files for use with ChemApp, FactSage, SimuSage, and ChemSheet come in a number of formats (see Thermodynamic data). Of these formats, only thermochemical data-files in ASCII format can be read and edited by the user.
Before you start editing a thermochemical data-file by hand, you might want to consider using a program called CSFAP offered by GTT-Technologies that provides an easy-to use graphical interface to edit, enhance, and merge data-files in ASCII format.
The basic structure of such a ChemApp data-file in ASCII format is shown in Table The basic structure of a ChemApp data-file. It comprises four sections:
- a header section
- data block(s) for mixture phases
- data block(s) for stoichiometric condensed phases
- a commentary block containing a text description of the data-file.
The appearance of the header section consists of six lines and is similar for all data-files. The content of the data blocks for mixture and stoichiometric condensed phases depends on the system under consideration. A commentary block can be added to the data-file after the last line of data. This can include, for example, comments, references to data sources, a quality code, limitations on the use of the data-file, etc.
Stoichiometry matrix
The stoichiometry matrix defines the relationship between the species of a system and the system components. It provides information important for the determination of the mass balances in a Gibbs energy minimisation calculation. Hence, it is of great significance in the successful compilation of a thermochemical data-file. Refer to Definition of a stoichiometry matrix for details on the structure of the stoichiometric matrix and guidance on its preparation.
The reference [Eri90] provides additional information.
HEADER
Six lines with a descriptive title and five lines of code containing general information
|
DATA BLOCK(S) FOR MIXTURE PHASES
Each block contains: phase name, model name; and the following information for all phase components - name, thermochemical data option, number of consecutive temperature ranges, stoichiometry coefficients, Gibbs energy data, and, if applicable, excess Gibbs energy data for
Mixture Phase 1
Mixture Phase 2
.
.
.
Mixture Phase 'n'
|
DATA BLOCK(S) FOR STOICHIOMETRIC CONDENSED PHASES
Stoichiometric Condensed Phase 1
Stoichiometric Condensed Phase 2
.
.
.
Stoichiometric Condensed Phase 'n'
|
COMMENTARY BLOCK
A text providing information on the origin of the data file, a quality designation, as well as a definition of the scope and areas of application of the data-file.
|
11.1 The format of a ChemApp data-file
The format of a ChemApp data-file is described in detail below, based on the sample data-file cosi.dat.
START OF HEADER SECTION
| Line 1: | Descriptive title
The title may be up to 78 characters long, and can be used to provide a brief description of, for example, the contents of the data-file and the source of the data. The title below describes the data-file cosi.dat supplied with ChemApp.
C-O-Si, also used for silicon arc furnace reactor calculations |
||||
|---|---|---|---|---|---|
| Line 2: | Code defining the size of the data-file
First digit: number of system components
Second digit: number of mixture phases
Third to last but one digit: number of phase constituents in the mixture phases
Last digit: number of stoichiometric condensed phases.
3 1 15 7 |
||||
| Line 3: | List of components in the system
The names of the components are listed, as shown. Each string must consist of 24 characters, including empty spaces.
C O Si |
||||
| Line 4: | Molecular weights of the system components. Use extra lines if appropriate. 12.0110 15.9994 28.0860 |
||||
| Line 5: | Definition of the temperature dependence terms for phase components and stoichiometric condensed phases in the Gibbs energy equation, G(T) = A + B*T + C*TlnT + D*T2 + E*T3 + F/T + (G*TU + H*TV + I*TW + J*TX + K*TY + L*TZ)
The equations can contain up to 12 terms. The first six terms are standard; once they are defined, they are valid for all substances contained in the data-file.
The last six terms (maximum) can be defined individually for each substance using real numbers, and thus derive separate Gibbs energy equations for each substance. The powers U to Z can have real number values between -9 and +9. Note: You can enter 99 for ln(T).
The entry in the data-file consists of the total number of standard terms defined, and their positions in the Gibbs energy equation. The code below for the cosi.dat data-file indicates that all of the first six terms are defined, and that the following equation applies, G(T) = A + B*T + C*T*lnT + D*T2 + E*T3 + F/T
6 1 2 3 4 5 6
|
||||
| Line 6: | Definition of the temperature and (if applicable) pressure dependence terms for the concentration coefficients in the excess Gibbs energy equation, L(T,P) = A + B*T + C*T*lnT + D*T2 + E*T3 + F/T + G*P + H*P2 Any number of the seven terms in the equation may be used. An entry consists of the total number of terms defined and their position in the Gibbs energy equation. For example, the entry below in the cosi.dat file defines the equation, L(T) = A 1 1 |
This ends the data entry for the header section of a data-file. The content of the six-line header for the data-file cosi.dat is summarised below.
HEADER SECTION OF THE DATA-FILE COSI.DAT
C-O-Si, also used for silicon arc furnace reactor calculations
3 1 15 7
C O Si
12.0110 15.9994 28.0860
6 1 2 3 4 5 6
1 1
END OF THE HEADER SECTION
START OF DATA BLOCK FOR MIXTURE PHASE(S)
Data are entered for all n mixture phases included in the system in any order sequence, except for the gas phase - this must always be considered as the first mixture phase. If a gas phase is not present, the number of its constituents must be set to 0. Data for each constituent of a mixture phase are entered successively, as shown below.
Data block for mixture phase 1 - the gas phase
| Entry 1: | The phase name
GAS
|
||
|---|---|---|---|
| Entry 2: | The model name - IDMX (ideal mixing)
IDMX
|
Data for component 1 of mixture phase 1 - carbon
| Entry 3: | The constituent name
C
|
|---|---|
| Entry 4: | Thermodynamic data option for constituent 1
In the following entry, first digit: thermodynamic data option (1 = Fixed-term Gibbs energy equations, see Table Available thermodynamic data options for other possible values), second digit: number of Gibbs energy equations, Remaining digits: coefficients of the stoichiometry matrix. These are entered in the order defined in line 3 of the header.
1 2 1.0 .0 .0 |
| Entry 5: | The upper temperature limit (K) and the temperature coefficients for each of the six terms in the Gibbs energy equation, G(T) = A + B*T + C*T*lnT +D*T2 + E*T3 + F/T
2000.0000 710457.00 -18.971944 -20.769400 -.27196000E-04
.00000000E+00 .00000000E+00
6000.0000 711696.00 -28.661644 -19.489100 -.35773200E-03
.00000000E+00 .00000000E+00
|
End of the data entry for component 1 of mixture phase 1
Subsequently, data are entered for the remaining phase constituents using the same procedure. The complete entry for the gas phase as the first mixture phase in the data-file cosi.dat is summarised in shortened form below.
DATA BLOCK FOR MIXTURE PHASE(S) IN COSI.DAT
GAS
IDMX
C
1 2 1.0 .0 .0
2000.0000 710457.00 -18.971944 -20.769400 -.27196000E-04
.00000000E+00 .00000000E+00
6000.0000 711696.00 -28.661644 -19.489100 -.35773200E-03
.00000000E+00 .00000000E+00
C2
1 1 2.0 .0 .0
6000.0000 826673.00 1.8085362 -30.685500 -.23765100E-02
.71128000E-07 -508774.00
C3
1 1 3.0 .0 .0
4000.0000 774485.00 130.77556 -49.887100 -.35584900E-02
.17642500E-06 602078.00
CO
1 1 1.0 1.0 .0
3000.0000 -119348.00 -5.9053738 -28.409400 -.20501600E-02
.00000000E+00 23012.000
CO2
1 1 1.0 2.0 .0
3000.0000 -409930.00 89.365756 -44.141200 -.45187200E-02
.00000000E+00 426768.00
O
1 2 .0 1.0 .0
2000.0000 243275.00 -21.813244 -20.874000 .25104000E-04
.00000000E+00 -48743.600
3000.0000 243168.00 -21.880544 -20.853100 .00000000E+00
.00000000E+00 .00000000E+00
:
:
:
Si
1 1 .0 .0 1.0
3000.0000 445338.00 -36.090244 -19.815400 -.50208000E-03
.00000000E+00 -100416.00
:
:
SiO
1 2 .0 1.0 1.0
2273.0000 -110346.00 -8.3075638 -29.823600 -.41191500E-02
.38004700E-06 103136.00
6000.0000 -130585.00 101.41956 -44.358800 .14267400E-02
-.62760000E-07 6301100.0
SiO2
1 1 .0 2.0 1.0
6000.0000 -325105.00 154.83356 -56.157600 -.15187900E-02
.58576000E-07 604170.00
END OF DATA BLOCK FOR MIXTURE PHASE(S)
DATA BLOCK FOR STOICHIOMETRIC CONDENSED PHASES
The basic data format for each stoichiometric condensed phase is the same as that for the components of a mixture phase. The entry for carbon as a stoichiometric condensed phase is shown below. The entry for pure silicon in this example is different. The data are entered in the alternative form, based on the use of a four-term heat capacity equation and explicit values for Delta H298 and S298.
| Note: | If different structural varieties of the same stoichiometric composition are to be treated explicitly, they must be given as separate phases. |
|---|
Data entry for a stoichiometric condensed phase - carbon
C
1 3 1.0 .0 .0
800.0000 -1153.5000 -12.583000 3.0258000 -.23742000E-01
.39214000E-05 22809.000
1900.0000 -12380.000 139.22000 -20.022000 -.24390000E-02
.19766000E-06 1066600.0
6000.0000 -17431.000 178.95000 -25.499000 -.98209000E-04
-.15854000E-07 1897800.0
Data entry for a stoichiometric condensed phase - pure silicon
The data for pure silicon are entered using a four-term heat capacity equation and explicit data for the enthalpy of formation (Delta H298), the entropy (S298), and temperatures and enthalpies of transformation. The heat capacity equation has the form Cp = a + b*T + c*T2 + d/T2
| Note: | Heat capacity data may be entered only if the Gibbs energy equations are defined as in line 5 of the header section, i.e. 6 1 2 3 4 5 6. |
|---|---|
| Note: | For pure silicon, both the solid and liquid state are included in the same set of Gibbs energy data. |
| Note: | Transformation enthalpies are entered for all transition temperature inputs, except the last. |
| Entry 1: | Phase name
Si
|
| Entry 2: | Thermodynamic data option (7 = enthalpy, entropy, and fixed-term heat capacity equations, see Table Available thermodynamic data options for other possible values), number of data inputs (i.e. temperature ranges for which specific heat data are available), coefficients of the stoichiometry matrix.
7 2 .0 .0 1.0
|
| Entry 3: | Enthalpy in J.mol-1 at 298.15 K, and entropy in J.K-1mol-1 at 298.15 K.
.00000000E+00 18.819600
|
| Entry 4: | Upper temperature limit in Kelvin, the four coefficients of the heat capacity equation (a, b, c, and d) applicable at temperatures up to the maximum temperature, Tmax(1).
Tmax(1) a b c d
1685.0000 22.823720 .38576480E-02 .00000000E+00 -353966.40
|
| Entry 5: | Delta H(trans1) at Tmax(1)
50208.000
|
| Entry 6: | Same input as for Entry 4, for Tmax(1)<T<Tmax(2)
Tmax(2) a b c d
3492.0000 27.196000 .00000000E+00 .00000000E+00 .00000000E+00
No transformation enthalpy is entered here, since this is the last data range. |
Data entry for a stoichiometric condensed phase - SiC
SiC
1 1 1.0 .0 1.0
3259.0000 -91845.000 280.43000 -42.593100 -.41798200E-02
.21198900E-06 829687.00
Data entry for a stoichiometric condensed phase - SiO2(quartz)
In this case, three extra terms in the Gibbs energy equation are applied, as discussed in the remarks to line 5 above.
| Entry 1: | Phase name
SiO2(quartz)
|
|---|---|
| Entry 2: | Thermodynamic data option (4 = Extended Gibbs energies, see Table Available thermodynamic data options for other possible values), number of Gibbs energy equations, and coefficients of the stoichiometry matrix.
4 3 .0 2.0 1.0
|
| Entry 3: | Upper temperature limit in Kelvin and temperature coefficients of the first Gibbs energy equation in Joule (see line 5)
373.00000 -935388.53 536.02533 -80.011990 .00000000E+00
.00000000E+00 1773342.0
|
| Entry 4: | Additional terms used
First digit: 3 = number of additional coefficients
Remaining numbers: 3 coefficient/power pairs
3 .00000000E+00 4.0 -961.10400 .5 -81928064. -2.0
|
| Entry 5: | Upper temperature limit in Kelvin and temperature coefficients of the second Gibbs energy equation in Joule (see line 5)
848.00000 -935486.57 537.07568 -80.011990 -.42200108E-02
.75354500E-05 1773342.0
|
| Entry 6: | Additional terms used
First digit: 3 = number of additional coefficients
Remaining numbers: 3 coefficient/power pairs
3 .50458704E-08 4.0 -961.10400 .5 -81928064. -2.0
|
| Entry 7: | Upper temperature limit in Kelvin and temperature coefficients (see line 5) of the third Gibbs energy equation in Joule
1744.2500 -933315.35 533.27853 -80.011990 .00000000E+00
.00000000E+00 1773342.0
|
| Entry 8: | Additional terms used
First digit: 3 = number of additional coefficients
Remaining numbers: 3 coefficient/power pairs
3 .00000000E+00 4.0 -961.10400 .5 -81928064. -2.0
|
End of data entry for SiO2(quartz)
Note: For the above entries, the extra terms do not have to use the same powers in each range. It is a coincidental in this example. All other phases of SiO2 are treated in the same way as SiO2(quartz). The data block is summarised below.
DATA BLOCK FOR STOICHIOMETRIC CONDENSED PHASES IN COSI.DAT
C
1 3 1.0 .0 .0
800.00000 -1153.5000 -12.583000 3.0258000 -.23742000E-01
.39214000E-05 22809.000
1900.0000 -12380.000 139.22000 -20.022000 -.24390000E-02
.19766000E-06 1066600.0
6000.0000 -17431.000 178.95000 -25.499000 .98209000E-04
-.15854000E-07 1897800.0
Si
7 2 .0 .0 1.0
.00000000E+00 18.819600
1685.0000 22.823720 .38576480E-02 .00000000E+00 -353966.40
50208.000
3492.0000 27.196000 .00000000E+00 .00000000E+00 .00000000E+00
SiC
1 1 1.0 .0 1.0
3259.0000 -91845.000 280.43000 -42.593100 -.41798200E-02
.21198900E-06 829687.00
SiO2(quartz)
4 3 .0 2.0 1.0
373.00000 -935388.53 536.02533 -80.011990 .00000000E+00
.00000000E+00 1773342.0
3 .00000000E+00 4.0 -961.10400 .5 -81928064. -2.0
848.00000 -935486.57 537.07568 -80.011990 -.42200108E-02
.75354500E-05 1773342.0
3 -.50458704E-08 4.0 -961.10400 .5 -81928064. -2.0
1744.2500 -933315.35 533.27853 -80.011990 .00000000E+00
.00000000E+00 1773342.0
3 .00000000E+00 4.0 -961.10400 .5 -81928064. -2.0
SiO2(tridymite)
4 2 .0 2.0 1.0
390.15000 -946102.84 500.41936 -75.372670 -.91038535E-01
.20563620E-03 2979047.5
3 -.17418275E-06 4.0 .00000000E+00 .5 -.15970769E+09 -2.0
1991.2800 -944111.17 480.75284 -75.372670 .00000000E+00
.00000000E+00 2979047.5
3 .00000000E+00 4.0 .00000000E+00 .5 -.15970769E+09 -2.0
SiO2(cristobalite)
4 3 .0 2.0 1.0
535.15000 -926547.67 571.80290 -83.513600 -.10104998E-01
.20917698E-04 1227680.0
3 -.16237635E-07 4.0 -1498.7720 .5 -46678699. -2.0
1995.9900 -924997.14 566.99971 -83.513600 .00000000E+00
.00000000E+00 1227680.0
3 .00000000E+00 4.0 -1498.7720 .5 -46678699. -2.0
3000.0000 -961789.83 569.34943 -85.772000 .00000000E+00
.00000000E+00 .00000000E+00
3 .00000000E+00 4.0 .00000000E+00 .5 .00000000E+00 -2.0
SiO2(liquid)
4 2 .0 2.0 1.0
1995.9900 -915415.78 562.19940 -83.513600 .00000000E+00
.00000000E+00 1227680.0
3 .00000000E+00 4.0 -1498.7720 .5 -46678699. -2.0
3000.0000 -952208.47 564.54913 -85.772000 .00000000E+00
.00000000E+00 .00000000E+00
3 .00000000E+00 4.0 .00000000E+00 .5 .00000000E+00 -2.0
END OF DATA BLOCK FOR STOICHIOMETRIC CONDENSED PHASES
Data-file commentary
Date : Nov.1992
Originator : KH+GE
Serial No : COSI.DAT
Quality Status : A
ChemSage Vers. : 4.0
Temperatures : RT to 3500K
Compositions : All
Applications : Phase equilibria and thermodynamic properties.
This files serves as an example of input for several type
of Gibbs energy data of pure substances, e.g. simple
G-functions with several temperature ranges (Type 1), H-S-Cp
input with implicit phase transformation (Type 7), extended
G-functions with several temperature ranges (Type 4).
Sources of data
Unary : THERDAS (90Spe); silicates from 85Ber
Binary : None
References : 85Ber Berman,R.G.; Brown,T.H.; Greenwood,H.J.: Atomic
Energy of Canada Limited, TR-377, 1985
90Spe Spencer,P.J.; Hack,K. : Swiss Materials 2,3a,
1990, p69
If you prepare your own data-files, we recommend that these be documented in a similar manner to that described above for GTT data-files, providing, for example, source(s) of the data, literature references, and other relevant information.
| 1 | Fixed-term Gibbs energies |
|---|---|
| 2 | Fixed-term Gibbs energies, constant molar volumes |
| 3 | Fixed-term Gibbs energies, P,T-dependent molar volumes |
| 4 | Extended Gibbs energies |
| 5 | Extended Gibbs energies, constant molar volumes |
| 6 | Extended Gibbs energies, P,T-dependent molar volumes |
| 7 | Enthalpy, entropy, fixed-term heat capacities |
| 8 | Enthalpy, entropy, fixed-term heat capacities, constant molar volumes |
| 9 | Enthalpy, entropy, fixed-term heat capacities, P,T-dependent molar volumes |
| 10 | Enthalpy, entropy, extended heat capacities |
| 11 | Enthalpy, entropy, extended heat capacities, constant molar volumes |
| 12 | Enthalpy, entropy, extended heat capacities, P,T-dependent molar volumes |
11.2 Data-files with preset suppression of species
In some cases it might be chosen to suppress phases or phase constituents from calculations (i.e. set their status to 'DORMANT', see Table Possible statuses for phases and constituents).
When a miscibility gap exists in the system
In such cases, the phase which exhibits the miscibility gap is contained twice in the data-file, but used only once in a normal phase equilibrium calculation. Hence, the second entry of this phase is suppressed from normal calculations by marking it with an exclamation mark ('!') in the 26th column of the data-file, see above.
| Note: | When calculations are performed in composition and temperature regions close to the miscibility gap, the second phase must be re-entered during run time by calling the subroutine TQCSP. |
|---|
When a data-file does not cover the entire composition range of a system
This is almost always the case when one of the phases has been described with a dilute solution approximation. In such cases, the file may contain phases, which are not stable under many conditions. These phases are suppressed in the data-file in order to speed up the calculations. However, it is important to monitor the results for the activities of these phases in the equilibrium output table, and to re-enter the phase and repeat the calculations if activity values greater than one are found.
GTT-Technologies may sometimes supply standard or customised data-files with suppressed species; which species have been suppressed and the reason why will be described in the commentary section.
If phases or phase constituents are to be assigned the status 'ELIMINATED' (see Table Possible statuses for phases and constituents), this can also be preset in the data-file. In this case a double cross ('#') is placed in the 26th column of the line containing the name of the phase or phase constituent in question. In both cases, the status can always be changed during run time using TQCSP and TQCSPC.
11.3 Definition of a stoichiometry matrix
The stoichiometry matrix defines the compositional relationship between the species of a system and the system components. The number of mols of each element in any species is presented in a fixed, repeatable form. A dataset for the specific species is formed by adding thermodynamic data (Gibbs energy coefficients) to the stoichiometry matrix.
In a stoichiometry matrix, each species is defined by a line, and each component by a column. For example, consider a system with the independent components Cr and C, in which the solid compound Cr23C6 exists. The stoichiometry matrix for 1 mol of Cr23C6 is
C Cr
Cr23C6 6 23
If a third element is present, e.g. Fe, an additional column is added, and the stoichiometric matrix for 1 mol of Cr23C6 becomes
C Cr Fe
Cr23C6 6 23 0
Sometimes it is more convenient to refer to 1 mol of atoms in the phase, the matrix then has the following form:
C Cr Fe
(Cr23C6)/29 0.2069 0.7931 0
Stoichiometry Matrix Plus Data Equals Dataset
The following example demonstrates the entry of the stoichiometry matrix with Gibbs energy coefficients to form a dataset for Cr23C6 for a system containing only C and Cr.
Cr23C6
1 1 6.0 23.0
1823.0000 -655776.00 4251.6000 -707.76500 -.89244700E-01
0.00000000 6052160.0
If more than two components are contained in a system, zeros are added to the stoichiometric matrix to represent the presence of the other components. This is shown in the next example, in which C is the second and Cr the fourth system component in a system containing a total of eleven components:
(Cr23C6)/29
1 1 0.0 .2069 0.0 .7931 0.0 0.0 0.0 0.0 0.0 0.0 0.0
1823.0000 -22613.000 146.60700 -24.405700 -.30774000E-02
0.00000000 208695.00
The example above also demonstrates the use of the matrix in reference to 1 mol of atoms. Note that any scaling of the stoichiometric coefficients of a species, caused by this type of entry, must also be accompanied by an identical scaling of the Gibbs energy coefficients. In addition, in order to avoid possible confusion when reading the output tables, it is good practice to change the species name, as in this case to (Cr23C6)/29.
12 FORTRAN example code (cademo1.f)
The program cademo1.f whose source code is included below is part of every FORTRAN distribution of ChemApp. When receiving ChemApp it is useful to compile and run this program, and compare its results to the master results, which for most versions of ChemApp are included on the distribution disks, and which are also printed here. The program's main purpose is to call virtually every ChemApp routine to verify that the ChemApp installation is working properly. For implementations of this program in languages other than FORTRAN (see for instance C example code (cademo1.c)) it also serves to verify the interface to ChemApp in this language. In contrast to the other code examples in this manual it also demonstrates how every ChemApp subroutine should be checked for the value of the error variable it returns, and act accordingly if it indicates an error. If you are using ChemApp with a language other than FORTRAN or C, your distribution disk contains a program similar to this one, even tough it might not be included in the appendix of this manual.
! Note that this program contains phase target calculations, ! which cannot be performed with the 'light' version of ChemApp ! Please also read the notes regarding the use of ChemApp with FORTRAN, ! which are contained in a separate file. ! ! Please direct any questions about this program or the results it ! produces on your machine to: ! ! GTT-Technologies ! Kaiserstrasse 100 ! 52134 Herzogenrath ! Germany ! ! Phone: +49-2407-59533 ! Fax: +49-2407-59661 ! E-mail: support@gtt-technologies.de ! WWW: http://www.gtt-technologies.de/ ! --------------------------------------------------------------------- ! Subroutine : CABORT ! --------------------------------------------------------------------- ! Subject : If a ChemApp error occurs, this function reports the error ! number and the routine it occurred in before it exits the ! program. ! ! --------------------------------------------------------------------- SUBROUTINE CABORT(LINENO, SRNAME, NOERR) IMPLICIT NONE INTEGER LINENO CHARACTER SRNAME*(*) INTEGER NOERR 41 FORMAT(1X,A,I4,2A) WRITE(UNIT=*,FMT=41) 'ChemApp error no. ',NOERR, * ' occurred when calling ', srname WRITE(UNIT=*,FMT=41) 'Aborting on line ',LINENO,' of ', 'CADEMO1' STOP END ! --------------------------------------------------------------------- ! Function: STRLEN ! --------------------------------------------------------------------- ! Subject : The support function STRLEN returns the total number of ! characters in a string, not counting trailing blanks ! ! --------------------------------------------------------------------- INTEGER FUNCTION STRLEN(INSTR) CHARACTER INSTR*(*) STRLEN = LEN(INSTR) DO WHILE(INSTR(STRLEN:STRLEN) .EQ. ' ') STRLEN = STRLEN - 1 IF (STRLEN .EQ. 1) RETURN ENDDO RETURN END PROGRAM CADEMO IMPLICIT NONE ! Declaration of variables to be used in the program ! Declarations of integer variables for use with ChemApp: ! Declaring the return variable for the function STRLEN INTEGER STRLEN ! An all-purpose integer variable INTEGER I ! Integer variables which hold the index number of phases, phase ! constituents, and sublattices INTEGER INDEXP, INDEXC, INDEXL ! The standard error code return variable INTEGER NOERR ! A variable that is set to the version number of ChemApp INTEGER CAVERS ! This variable is used in conjunction with TQLITE to determine whether ! the program is linked to the regular or the 'light' version of ! ChemApp. In the latter case, target calculations are left out. INTEGER ISLITE ! A number of integer variables to return the information from TQSIZE INTEGER LIA,LIB,LIC,LID,LIE,LIF,LIG,LIH,LII,LIJ,LIK ! A similar set of integer variables to return the information from ! TQUSED INTEGER LDIA,LDIB,LDIC,LDID,LDIE,LDIF,LDIG,LDIH,LDII,LDIJ,LDIK ! An integer that will contain the index number of a condition set INTEGER NUMCON ! An integer that will contain the number of phases in the data-file ! loaded INTEGER NPHASE ! An integer that will hold the number of phase constituents in the gas ! phase INTEGER NPCGAS ! An integer that will contain the number of system components INTEGER NSCOM ! Integers that will contain the number of sublattices, and the number of ! sublattice contituents of a particular sublattice INTEGER NSL, NSLCON ! Two integers used for one-dimensional phase mapping calculations, one ! to indicate whether we need to make more calls to TQMAP/TQMAPL INTEGER ICONT ! And another one to keep track of the number of results we obtained INTEGER RESNO ! An integer that holds the FORTRAN unit number under which the ! thermochemical data-files will be opened INTEGER UNITNO ! An integer that holds the FORTRAN unit number to which the ! ChemApp error messages are written by default INTEGER EUNIT ! Variables to store information from various transparent file header ! fields INTEGER TFHVER, TFHVNW(3), TFHVNR(3), TFHDTC(6), TFHDTE(6) ! These integers will be used to store the HASP dongle id, plus the ! ChemApp expiration month and year INTEGER HASPID, EDMON, EDYEAR ! Declarations of real variables for use with ChemApp: ! An all-purpose real variable DOUBLE PRECISION D1 ! An array of two real variables... DOUBLE PRECISION DARR2(2) ! ...and one of 30 DOUBLE PRECISION DARR30(30) ! A variable to hold the molecular mass of a system component (see ! TQSTSC) or a phase constituent (see TQSTPC) DOUBLE PRECISION WMASS ! This particular array of two real variables will contain the ! temperature and pressure of a stream to be defined with TQSTTP DOUBLE PRECISION TP(2) ! Declarations of string variables for use with ChemApp: ! Every 'name' used in conjuction with ChemApp, whether it is the name ! of a phase, a system component, a phase constituent, or a stream, can ! be up to 24 characters in length. Exceptions are the subroutine TQERR, ! as well as the subroutines TQGTRH, TQGTID, TQGTNM, TQGTPI, and TQWSTR, ! which all pass or return strings longer than 24 characters. ! An all-purpose string for ChemApp CHARACTER DSTR*24 ! Character strings that will hold names of phases, phase constituents, ! system components, mixture model names, as well as sublattices and ! their constituents CHARACTER*24 PNAME, CNAME, MNAME ! The following variable is an array of 3 strings. It will be used with ! TQCSC, which is the only ChemApp subroutine that takes a character ! array as input. CHARACTER NEWSC(3)*24 ! The following character array will hold a ChemApp error message. In ! this case, it will be used to retrieve the copyright string. CHARACTER MESS(3)*80 ! The next three character strings are used to hold information ! about the copy of ChemApp used. CHARACTER ID*255, NAME*80, PID*24 ! Variables to store information from various transparent file header ! fields CHARACTER TFHID*255, TFHUSR*80,TFHNWP*40,TFHNRP*40,TFHREM*80 ! This character variable will be used to store the HASP dongle type CHARACTER HASPT*5 ! Format declarations 51 FORMAT(1X,A) 52 FORMAT(1X,A,I4) 53 FORMAT(1X,A,//) 54 FORMAT(1X,A,I4,//) 55 FORMAT(1X,//,1X,A,//) 56 FORMAT(1X,A,I3,A,I3,A) 57 FORMAT(1X,A,G12.5) 58 FORMAT(1X,A,G12.5,//) 59 FORMAT(1X,A,F8.2,A) 60 FORMAT(1X,A,I3,A) 61 FORMAT(1X,A,I4.4,5(A1,I2.2)) 62 FORMAT(1X,A,I2,A,I4,//) 63 FORMAT(1X,A,I12,//) ! Print the program title WRITE(UNIT=*,FMT=51) 'This is cademo1, a FORTRAN program ' // * 'to test ChemApp and to demonstrate ' WRITE(UNIT=*,FMT=53) 'the use of every ChemApp subroutine.'
This is cademo1, a FORTRAN program to test ChemApp and to demonstrate the use of every ChemApp subroutine.
! Initialise ChemApp CALL TQINI(NOERR) ! After all following invocations of ChemApp subroutines, the error ! variable will be checked for a non-zero value, in which case an ! error has occurred and the program is aborted IF (NOERR .NE. 0) CALL CABORT(225,'TQINI',NOERR) ! Print the copyright string. The way to do this is to first ! call the subroutine TQCPRT, which copies the copyright string into ! the ChemApp-internal error buffer string: CALL TQCPRT(NOERR) IF (NOERR .NE. 0) CALL CABORT(232,'TQCPRT',NOERR) ! Then do what is usually done to retrieve an error message: ! call TQERR to retrieve it. CALL TQERR(MESS,NOERR) IF (NOERR .NE. 0) CALL CABORT(237,'TQERR',NOERR) ! To print out the message, print all the three strings of ! MESS : WRITE(UNIT=*,FMT='(3(A,/))') MESS(1), MESS(2), MESS(3)
This program contains ChemApp Copyright GTT-Technologies, Kaiserstrasse 100, D-52134 Herzogenrath, Germany http://www.gtt-technologies.de
! Get version number of ChemApp CALL TQVERS(CAVERS,NOERR) IF (NOERR .NE. 0) CALL CABORT(247,'TQVERS',NOERR) WRITE(UNIT=*,FMT=54) 'ChemApp version is: ', CAVERS ! Get array sizes CALL TQSIZE(LIA,LIB,LIC,LID,LIE,LIF,LIG,LIH,LII,LIJ,LIK,NOERR) IF (NOERR .NE. 0) CALL CABORT(252,'TQSIZE',NOERR) WRITE(UNIT=*,FMT=51) 'Internal array sizes of this version '// * 'of ChemApp,' WRITE(UNIT=*,FMT=51) 'maximum number of:' WRITE(UNIT=*,FMT=52) 'constituents: ',LIA WRITE(UNIT=*,FMT=52) 'system components: ',LIB WRITE(UNIT=*,FMT=52) 'mixture phases: ',LIC WRITE(UNIT=*,FMT=52) 'excess Gibbs energy '// * 'coefficients for a mixture phase: ',LID WRITE(UNIT=*,FMT=52) 'excess magnetic '// * 'coefficients for a mixture phase: ',LIE WRITE(UNIT=*,FMT=52) 'sublattices for a '// * 'mixture phase: ',LIF WRITE(UNIT=*,FMT=52) 'constituents of a '// * 'sublattice: ',LIG WRITE(UNIT=*,FMT=51) 'oxide constituents of a phase described '// * 'by the Gaye-Kapoor-Frohberg ' WRITE(UNIT=*,FMT=52) ' or modified quasichemical '// * 'formalisms: ',LIH WRITE(UNIT=*,FMT=52) 'Gibbs energy/heat '// * 'capacity equations for a constituent: ',LII WRITE(UNIT=*,FMT=52) 'Gibbs energy/heat '// * 'capacity equations: ',LIJ WRITE(UNIT=*,FMT=54) 'constituents with '// * 'P,T-dependent molar volumes: ',LIK
ChemApp version is: 645 Internal array sizes of this version of ChemApp, maximum number of: constituents: 1600 system components: 48 mixture phases: 44 excess Gibbs energy coefficients for a mixture phase: 1999 excess magnetic coefficients for a mixture phase: 499 sublattices for a mixture phase: 5 constituents of a sublattice: 1600 oxide constituents of a phase described by the Gaye-Kapoor-Frohberg or modified quasichemical formalisms: 25 Gibbs energy/heat capacity equations for a constituent: 20 Gibbs energy/heat capacity equations: 4800 constituents with P,T-dependent molar volumes: 1600
! Get value for input/output option ! Determine which FORTRAN unit is used by default by TQRFIL for ! reading the thermochemical data-file, and use the returned number to ! subsequently open and close the data-file CALL TQGIO('FILE ', UNITNO, NOERR) IF (NOERR .NE. 0) CALL CABORT(286,'TQGIO',NOERR) WRITE(UNIT=*,FMT=54) 'The thermochemical data will be read '// * 'from the file associated with unit ', UNITNO
The thermochemical data will be read from the file associated with unit 10
! Open ASCII data-file for reading CALL TQOPNA('cosi.dat', UNITNO, NOERR) IF (NOERR .NE. 0) CALL CABORT(294,'TQOPNA',NOERR) ! Read data-file CALL TQRFIL(NOERR) IF (NOERR .NE. 0) CALL CABORT(298,'TQRFIL',NOERR) ! Close data-file CALL TQCLOS(UNITNO, NOERR) IF (NOERR .NE. 0) CALL CABORT(302,'TQCLOS',NOERR) ! TQUSED is used to find out to what extent the thermochemical data ! storage space in the internal arrays of ChemApp (as reported by ! TQSIZE) are occupied by the currently loaded thermochemical system. CALL TQUSED(LDIA,LDIB,LDIC,LDID,LDIE,LDIF,LDIG,LDIH,LDII, * LDIJ,LDIK,NOERR) IF (NOERR .NE. 0) CALL CABORT(310,'TQUSED',NOERR) ! As an example, check how many Gibbs energy/heat capacity equations ! are currently being used. WRITE(UNIT=*,FMT=52) 'Actual number of Gibbs energy/heat ' // * 'capacity equations used (TQUSED):', LDIJ WRITE(UNIT=*,FMT=52) 'Maximum number in this version ' // * 'of ChemApp (TQSIZE):', LIJ
Actual number of Gibbs energy/heat capacity equations used (TQUSED): 35 Maximum number in this version of ChemApp (TQSIZE):4800
! Get system units CALL TQGSU('Pressure ',DSTR,NOERR) IF (NOERR .NE. 0) CALL CABORT(323,'TQGSU',NOERR) WRITE(UNIT=*,FMT=51) 'Pressure unit: ' // DSTR CALL TQGSU('Volume ',DSTR,NOERR) IF (NOERR .NE. 0) CALL CABORT(327,'TQGSU',NOERR) WRITE(UNIT=*,FMT=51) 'Volume unit: ' // DSTR CALL TQGSU('Temperature ',DSTR,NOERR) IF (NOERR .NE. 0) CALL CABORT(331,'TQGSU',NOERR) WRITE(UNIT=*,FMT=51) 'Temperature unit: ' // DSTR CALL TQGSU('Energy ',DSTR,NOERR) IF (NOERR .NE. 0) CALL CABORT(335,'TQGSU',NOERR) WRITE(UNIT=*,FMT=51) 'Energy unit: ' // DSTR ! Change system unit ! Here we change the unit for the quantity 'amount' to gram, ! mainly so that when we call TQSTSC further down, we get the ! molecular mass expressed in the unit g/mol CALL TQCSU('Amount ','gram ', NOERR) IF (NOERR .NE. 0) CALL CABORT(343,'TQCSU',NOERR) WRITE(UNIT=*,FMT=53) 'Amount unit set to gram'
Pressure unit: bar Volume unit: dm3 Temperature unit: K Energy unit: J Amount unit set to gram
! Get number of system components CALL TQNOSC(NSCOM,NOERR) IF (NOERR .NE. 0) CALL CABORT(351,'TQNOSC',NOERR) WRITE(UNIT=*,FMT=52) 'Number of system components: ',NSCOM ! Get stoichiometry of system component ! Note that the array used (DARR30) is much bigger than needed, ! which doesn't matter to ChemApp, as long as it is big enough. CALL TQSTSC(1,DARR30,WMASS,NOERR) IF (NOERR .NE. 0) CALL CABORT(359,'TQSTSC',NOERR) ! Here the array elements are printed, which describe the stoichiometry ! of the system component. This particular piece of information is ! not too useful though, since the array contains all zeros, except ! for a '1.0' at the position of the system component we specified. WRITE(UNIT=*,FMT='(1X,A,3F4.1)') 'Stoichiometry of system '// * 'component 1: ', * DARR30(1), DARR30(2), DARR30(3) ! Had we not changed the amount unit from the default value 'mol' to ! 'gram', we would receive a value of 1.0 for wmass. The reason is that ! the molecular mass is always output using the unit [current amount ! unit]/mol. If the current amount unit happens to be mol, the value for ! wmass will be 1.0. WRITE(UNIT=*,FMT=57) 'Molecular mass in g/mol: ', WMASS ! Get name of system component #1 CALL TQGNSC(1,DSTR,NOERR) IF (NOERR .NE. 0) CALL CABORT(379,'TQGNSC',NOERR) WRITE(UNIT=*,FMT=51) 'Name of system component 1: ' // DSTR ! Get index number of system component ! This is the reverse of the above, using the name of the ! system component to get its index number CALL TQINSC(DSTR, I, NOERR) IF (NOERR .NE. 0) CALL CABORT(386,'TQINSC',NOERR) WRITE(UNIT=*,FMT=54) 'Index number of system component '// * DSTR // ' is: ', I
Number of system components: 3 Stoichiometry of system component 1: 1.0 0.0 0.0 Molecular mass in g/mol: 12.011 Name of system component 1: C Index number of system component C is: 1
! Change system components ! TQCSC is the only ChemApp subroutine which takes a character ! array as input. The array contains as many strings as there ! are system components in the data-file. The system components are ! changed to SiO, SiC, and CO. NEWSC(1) = 'SiO' NEWSC(2) = 'SiC' NEWSC(3) = 'CO' CALL TQCSC(NEWSC,NOERR) IF (NOERR .NE. 0) CALL CABORT(403,'TQCSC',NOERR) WRITE(UNIT=*,FMT=51) 'System components changed to SiO-SiC-CO.' ! Print names of new system components DO I=1, NSCOM CALL TQGNSC(I,DSTR,NOERR) IF (NOERR .NE. 0) CALL CABORT(412,'TQGNSC',NOERR) WRITE(UNIT=*,FMT='(1X,A,I4,A)') 'Name of new system '// * 'component ', I, ': '// DSTR ENDDO ! Get stoichiometry of system component #1 CALL TQSTSC(1,DARR30,D1,NOERR) IF (NOERR .NE. 0) CALL CABORT(419,'TQSTSC',NOERR) ! Note that the stoichiometry of the system component #1 does not ! change, as compared to the first call to TQSTSC before the ! system components have been altered: WRITE(UNIT=*,FMT='(1X,A,3F4.1)') 'Stoichiometry of system '// * 'component 1: ', * DARR30(1), DARR30(2), DARR30(3) ! What changes of course is the molecular mass: WRITE(UNIT=*,FMT=58) 'Molecular mass in g/mol: ', D1
System components changed to SiO-SiC-CO. Name of new system component 1: SiO Name of new system component 2: SiC Name of new system component 3: CO Stoichiometry of system component 1: 1.0 0.0 0.0 Molecular mass in g/mol: 44.085
! Get number of phases CALL TQNOP(NPHASE,NOERR) IF (NOERR .NE. 0) CALL CABORT(437,'TQNOP',NOERR) WRITE(UNIT=*,FMT=52) 'Number of phases: ',NPHASE ! Get name of phase #1 CALL TQGNP(1,DSTR,NOERR) IF (NOERR .NE. 0) CALL CABORT(443,'TQGNP',NOERR) WRITE(UNIT=*,FMT=51) 'Name of phase 1: ' // DSTR ! Get index number of phase ! This is the reverse of the above, using the name of the ! phase to get its index number CALL TQINP(DSTR,I,NOERR) IF (NOERR .NE. 0) CALL CABORT(451,'TQINP',NOERR) WRITE(UNIT=*,FMT=52) 'Index number of phase ' // DSTR // * ' is: ', I ! Get model name of phase CALL TQMODL(1, DSTR, NOERR) IF (NOERR .NE. 0) CALL CABORT(458,'TQMODL',NOERR) WRITE(UNIT=*,FMT=51) 'Model name of phase 1: ' // DSTR ! Whereas above we asked for the model name of the first phase in ! the data-file, which is normally a mixture phase (gas phase), ! we now ask for the model name of the last phase in the ! data-file (NPHASE, as determined by a call to TQNOP). This is ! typically a stoichiometric condensed phase, in which case the ! string 'PURE' is returned as the model name CALL TQMODL(NPHASE, DSTR, NOERR) IF (NOERR .NE. 0) CALL CABORT(468,'TQMODL',NOERR) WRITE(UNIT=*,FMT='(1X,A,I2,A,//)') 'Model name of phase ', * nphase,': ' // DSTR ! Get number of phase constituents of phase #1 CALL TQNOPC(1, NPCGAS, NOERR) IF (NOERR .NE. 0) CALL CABORT(475,'TQNOPC',NOERR) WRITE(UNIT=*,FMT=52) 'Number of phase constituents in phase 1: ', * npcgas ! Get name of phase constituent CALL TQGNPC(1, 1, DSTR, NOERR) IF (NOERR .NE. 0) CALL CABORT(481,'TQGNPC',NOERR) WRITE(UNIT=*,FMT=51) 'Name of phase constituent 1 of phase 1: '// * dstr ! Get index number of phase constituent ! This is the reverse of the above, using the name of the phase ! constituent to get its index number CALL TQINPC(DSTR, 1, I, NOERR) IF (NOERR .NE. 0) CALL CABORT(490,'TQINPC',NOERR) WRITE(UNIT=*,FMT=52) 'Index number of phase constituent ' // * DSTR // ' is: ', I ! Check whether the phase constituent can be used as incoming species ! (note that there are only few cases, and only for certain mixture ! phase models, where this might not be permitted) CALL TQPCIS(1, 1, I, NOERR) IF (NOERR .NE. 0) CALL CABORT(498,'TQPCIS',NOERR) IF (I .EQ. 0) THEN WRITE(*,FMT=51) 'Usage of ' // DSTR // * ' as incoming species is not permitted.' ELSE WRITE(*,FMT=51) 'Usage of ' // DSTR // * ' as incoming species is o.k.' ENDIF ! Get stoichiometry of phase constituent ! TQSTPC is a subroutine similar to TQSTSC, except that with ! TQSTPC, the returned stoichiometry array contains valuable ! information. This stoichiometry array represents the ! stoichiometry matrix for a phase constituent, as described in ! the ChemApp manual CALL TQSTPC(1, 1, DARR30, WMASS, NOERR) IF (NOERR .NE. 0) CALL CABORT(514,'TQSTPC',NOERR) WRITE(UNIT=*,FMT='(1X,A,3F4.1)') 'Stoichiometry of phase '// * 'constituent 1 in phase 1 is: ', * DARR30(1), DARR30(2), DARR30(3) WRITE(UNIT=*,FMT=58) 'Molecular mass: ', WMASS
Number of phases: 8 Name of phase 1: GAS Index number of phase GAS is: 1 Model name of phase 1: IDMX Model name of phase 8: PURE Number of phase constituents in phase 1: 15 Name of phase constituent 1 of phase 1: C Index number of phase constituent C is: 1 Usage of C as incoming species is o.k. Stoichiometry of phase constituent 1 in phase 1 is: -0.5 0.5 0.5 Molecular mass: 12.011
! The system components are changed back to the original set of C, O, ! and Si, since we don't need the modified set of system components ! (SiO, SiC, and CO) any longer. NEWSC(1) = 'C' NEWSC(2) = 'O' NEWSC(3) = 'Si' CALL TQCSC(NEWSC,NOERR) IF (NOERR .NE. 0) CALL CABORT(530,'TQCSC',NOERR) WRITE(UNIT=*,FMT=53) 'System components changed back to C-O-Si.'
System components changed back to C-O-Si.
! Changing the status of phases and phase constituents ! Change status of phase #1 CALL TQCSP(1, 'eliminated ', NOERR) IF (NOERR .NE. 0) CALL CABORT(538,'TQCSP',NOERR) WRITE(UNIT=*,FMT=51) 'Status of phase 1 set to ''eliminated''' ! Get status of phase #1 CALL TQGSP(1, DSTR, NOERR) IF (NOERR .NE. 0) CALL CABORT(543,'TQGSP',NOERR) WRITE(UNIT=*,FMT=51) 'Status of phase 1 is: ' // DSTR ! Change status of phase #1 CALL TQCSP(1, 'entered ', NOERR) IF (NOERR .NE. 0) CALL CABORT(548,'TQCSP',NOERR) WRITE(UNIT=*,FMT=51) 'Status of phase 1 set to ''entered''' ! Get status of phase #1 CALL TQGSP(1, DSTR, NOERR) IF (NOERR .NE. 0) CALL CABORT(553,'TQGSP',NOERR) WRITE(UNIT=*,FMT=51) 'Status of phase 1 is: ' // DSTR
Status of phase 1 set to 'eliminated' Status of phase 1 is: ELIMINATED Status of phase 1 set to 'entered' Status of phase 1 is: ENTERED
! Change status of phase constituent #1 of phase #1 CALL TQCSPC(1, 1, 'dormant ', NOERR) IF (NOERR .NE. 0) CALL CABORT(560,'TQCSPC',NOERR) WRITE(UNIT=*,FMT=51) 'Status of constituent 1 of '// * 'phase 1 set to ''dormant''' ! Get status of phase constituent #1 of phase #1 CALL TQGSPC(1, 1, DSTR, NOERR) IF (NOERR .NE. 0) CALL CABORT(566,'TQGSPC',NOERR) WRITE(UNIT=*,FMT=51) 'Status of constituent 1 of ' // * 'phase 1 is: '//DSTR ! Change status of phase constituent #1 of phase #1 CALL TQCSPC(1, 1, 'entered ', NOERR) IF (NOERR .NE. 0) CALL CABORT(572,'TQCSPC',NOERR) WRITE(UNIT=*,FMT=51) 'Status of constituent 1 of phase 1 set '// * 'to ''entered''' ! Get status of phase constituent #1 of phase #1 CALL TQGSPC(1, 1, DSTR, NOERR) IF (NOERR .NE. 0) CALL CABORT(578,'TQGSPC',NOERR) WRITE(UNIT=*,FMT=53) 'Status of constituent 1 of ' // * 'phase 1 is: '//DSTR
Status of constituent 1 of phase 1 set to 'dormant' Status of constituent 1 of phase 1 is: DORMANT Status of constituent 1 of phase 1 set to 'entered' Status of constituent 1 of phase 1 is: ENTERED
WRITE(UNIT=*,FMT=53) 'Calculations using global conditions:'
! Set equilibrium conditions
! When setting equilibrium conditions with TQSETC, the currently
! set units for the various quantities (pressure, amount, etc.)
! apply. Thus, in the following call, the incoming amount ('ia')
! of phase constituent #4 of phase #1 is set to 1.0 gram.
CALL TQSETC('ia ', 1, 4, 1.D0,NUMCON,NOERR)
IF (NOERR .NE. 0) CALL CABORT(592,'TQSETC',NOERR)
! Change system unit
! Now we change the amount unit back to 'mol'. This does not
! affect any previously set values. The amount of phase
! constituent #4 of phase #1 we set above is still 1.0 gram, and
! is not changed to 1.0 mol!
CALL TQCSU('Amount ','mol ', NOERR)
IF (NOERR .NE. 0) CALL CABORT(600,'TQCSU',NOERR)
WRITE(UNIT=*,FMT=53) 'Amount unit set to mol'
Calculations using global conditions: Amount unit set to mol
! Since we changed the amount unit to 'mol', the call to TQSETC ! below inputs 3.0 mol of constituent #12 of phase #1 CALL TQSETC('ia ', 1, 12, 3.D0,NUMCON,NOERR) IF (NOERR .NE. 0) CALL CABORT(607,'TQSETC',NOERR) ! Now we input 8 mol of constituent #8 of phase #1 CALL TQSETC('ia ', 1, 8, 2.D0,NUMCON,NOERR) IF (NOERR .NE. 0) CALL CABORT(611,'TQSETC',NOERR) ! Remove equilibrium condition ! This call to TQREMC demonstrates how the parameter 'NUMCON' is ! used, which is returned with every call to TQSETC: The ! previously defined condition is removed again CALL TQREMC(NUMCON, NOERR) IF (NOERR .NE. 0) CALL CABORT(618,'TQREMC',NOERR) ! Set temperature ! Here TQSETC is used to enter the temperature. Since we didn't change ! the default temperature unit, it is still Kelvin ('K') CALL TQSETC('t ', 0, 0, 1800.D0,NUMCON,NOERR) IF (NOERR .NE. 0) CALL CABORT(624,'TQSETC',NOERR) ! Change limit of target variables ! The next call demonstrates how TQCLIM is used. Normally though ! calls to TQCLIM are only necessary under special circumstances CALL TQCLIM('plow ', 1.0D-49, NOERR) IF (NOERR .NE. 0) CALL CABORT(631,'TQCLIM',NOERR) ! Display present settings ! TQSHOW outputs the conditions which are active at this point WRITE(UNIT=*,FMT=51) 'Currently active conditions:' WRITE(UNIT=*,FMT=51) '**** Begin output table produced by TQSHOW' CALL TQSHOW(NOERR) WRITE(UNIT=*,FMT=53) '**** End output table produced by TQSHOW' IF (NOERR .NE. 0) CALL CABORT(640,'TQSHOW',NOERR)
Currently active conditions: **** Begin output table produced by TQSHOW TARGET LIMITS: Pressure/bar : 1.00000E-49 1.00000E+07 Volume/dm3 : 1.00000E-07 1.00000E+50 Temperature/K: 298.15 6000.00 T = 1800.00000000 K P = 1 bar STREAM CONSTITUENT CO/GAS/ AMOUNT/mol = 3.57010253334476E-02 Si3/GAS/ AMOUNT/mol = 3.00000000000000E+00 INACTIVE ELECTRON CONSTRAINT **** End output table produced by TQSHOW
! Calculate equilibrium ! A simple call to TQCE or TQCEL is sufficient to calculate the ! equilibrium. All input parameters to TQCE and TQCEL are only ! significant if target calculations are performed. DARR2(1)=0.0 CALL TQCE(' ',0,0,DARR2,NOERR) IF (NOERR .NE. 0) CALL CABORT(651,'TQCE',NOERR) ! Calculate and list equilibrium ! TQCEL calculates the equilibrium just like TQCE, with the only ! difference that a ChemSage-type result table is written to the ! file/unit associated with 'LIST' (see TQGIO/TQCIO) WRITE(UNIT=*,FMT=51) '**** Begin output table produced by TQCEL' CALL TQCEL(' ',0,0,DARR2,NOERR) WRITE(UNIT=*,FMT=53) '**** End output table produced by TQCEL' IF (NOERR .NE. 0) CALL CABORT(661,'TQCEL',NOERR)
**** Begin output table produced by TQCEL
T = 1800 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
CO/GAS/ 3.5701E-02
Si3/GAS/ 3.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
SiO 0.0000E+00 9.1172E-01 4.1405E-02
CO 0.0000E+00 8.8214E-02 4.0062E-03
Si 0.0000E+00 6.7137E-05 3.0490E-06
CO2 0.0000E+00 1.1335E-06 5.1478E-08
Si2C 0.0000E+00 7.0003E-07 3.1792E-08
Si2 0.0000E+00 4.5031E-07 2.0451E-08
SiO2 0.0000E+00 1.1236E-07 5.1026E-09
Si3 0.0000E+00 1.0170E-07 4.6188E-09
SiC 0.0000E+00 7.3913E-12 3.3567E-13
O 0.0000E+00 7.2737E-12 3.3033E-13
C 0.0000E+00 1.2567E-13 5.7072E-15
O2 0.0000E+00 1.6031E-16 7.2803E-18
C2 0.0000E+00 9.7381E-17 4.4225E-18
C3 0.0000E+00 4.0495E-17 1.8391E-18
O3 0.0000E+00 8.7034E-33 3.9526E-34
TOTAL: 0.0000E+00 1.0000E+00 4.5415E-02
mol ACTIVITY
Si 8.9464E+00 1.0000E+00
SiC 3.5701E-02 1.0000E+00
SiO2(cristobali) 1.7851E-02 1.0000E+00
SiO2(tridymite) 0.0000E+00 9.9970E-01
SiO2(liquid) 0.0000E+00 9.3907E-01
SiO2(quartz) T 0.0000E+00 9.1491E-01
C 0.0000E+00 2.2526E-02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
2.46520E+02 7.84578E+05 8.41507E+02 -7.30135E+05 0.00000E+00
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
**** End output table produced by TQCEL
! Once an equilibrium has been calculated using TQCE/TQCEL, subsequent ! calculations can be performed using TQCEN/TQCENL, which speeds up the ! equilibrium calculation by taking results from the previous ! calculation as initial estimates CALL TQSETC('t ', 0, 0, 1850.D0,NUMCON,NOERR) IF (NOERR .NE. 0) CALL CABORT(670,'TQSETC',NOERR) CALL TQCEN(' ', 0, 0, DARR2, NOERR) IF (NOERR .NE. 0) CALL CABORT(673,'TQCEN',NOERR) CALL TQSETC('t ', 0, 0, 1900.D0,NUMCON,NOERR) IF (NOERR .NE. 0) CALL CABORT(676,'TQSETC',NOERR) ! Similar to TQCEL, TQCENL also provides a ChemSage output table WRITE(UNIT=*,FMT=51) '**** Begin output table produced by TQCENL' CALL TQCENL(' ', 0, 0, DARR2, NOERR) WRITE(UNIT=*,FMT=53) '**** End output table produced by TQCENL' IF (NOERR .NE. 0) CALL CABORT(682,'TQCENL',NOERR) ! Change value of input/output option ! Redirect the output from TQCEL to a file using TQCIO with the ! option 'LIST'. ! The following call to TQCIO will redirect the output of ChemApp ! subroutines like TQCEL and TQSHOW to the FORTRAN unit number ! 21. Note that only certain values for the unit number are ! permitted (see the ChemApp manual entry for TQCIO). CALL TQCIO('LIST ', 21, NOERR) IF (NOERR .NE. 0) CALL CABORT(694,'TQCIO',NOERR) ! Note that at this point, no file has been associated with unit ! number 21 yet! This is done with the subsequent call to TQOPEN: CALL TQOPEN('result', 21, NOERR) IF (NOERR .NE. 0) CALL CABORT(700,'TQOPEN',NOERR) ! TQWSTR can be used to write user-defined text to the unit numbers ! associated with 'LIST' and 'ERROR'. This subroutine is especially ! useful for non-FORTRAN programs. CALL TQWSTR('LIST', 'Output from TQCEL (ChemSage result table):', * NOERR) IF (NOERR .NE. 0) CALL CABORT(707,'TQWSTR',NOERR) WRITE(UNIT=*,FMT=53) 'Output of next call to TQCEL will be '// * ' written to file ''result''' ! Now the ChemSage-type result table output by the following ! call to TQCEL is written to the file 'result': CALL TQCEL(' ',0,0,DARR2,NOERR) IF (NOERR .NE. 0) CALL CABORT(716,'TQCEL',NOERR) ! Close the file associated with unit number 21 again... CALL TQCLOS(21, NOERR) IF (NOERR .NE. 0) CALL CABORT(721,'TQCLOS',NOERR) ! ...and redirect the output of routines like TQCEL and TQSHOW ! back to unit number 6, which is the default and associated with ! the standard output unit: CALL TQCIO('LIST ', 6, NOERR) IF (NOERR .NE. 0) CALL CABORT(727,'TQCIO',NOERR) WRITE(UNIT=*,FMT=53) 'Output of subsequent calls to TQCEL/'// * 'TQSHOW/TQMAPL will go to UNIT 6 again'
**** Begin output table produced by TQCENL
T = 1900 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
CO/GAS/ 3.5701E-02
Si3/GAS/ 3.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
SiO 0.0000E+00 8.5125E-01 1.2301E-01
CO 0.0000E+00 1.4866E-01 2.1481E-02
Si 0.0000E+00 8.4025E-05 1.2142E-05
CO2 0.0000E+00 3.7718E-06 5.4503E-07
Si2C 0.0000E+00 1.4416E-06 2.0832E-07
Si2 0.0000E+00 7.4859E-07 1.0817E-07
SiO2 0.0000E+00 2.7664E-07 3.9975E-08
Si3 0.0000E+00 1.6036E-07 2.3173E-08
SiC 0.0000E+00 3.6804E-11 5.3182E-12
O 0.0000E+00 2.9557E-11 4.2711E-12
C 0.0000E+00 7.5814E-13 1.0955E-13
O2 0.0000E+00 1.3988E-15 2.0213E-16
C2 0.0000E+00 1.3301E-15 1.9220E-16
C3 0.0000E+00 7.2884E-16 1.0532E-16
O3 0.0000E+00 6.6967E-31 9.6770E-32
TOTAL: 0.0000E+00 1.0000E+00 1.4450E-01
mol ACTIVITY
Si 8.9464E+00 1.0000E+00
SiC 3.5701E-02 1.0000E+00
SiO2(cristobali) 1.7851E-02 1.0000E+00
SiO2(tridymite) 0.0000E+00 9.9918E-01
SiO2(liquid) 0.0000E+00 9.7125E-01
SiO2(quartz) T 0.0000E+00 9.0635E-01
C 0.0000E+00 3.4700E-02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
2.46541E+02 8.09231E+05 8.54837E+02 -8.14958E+05 0.00000E+00
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
**** End output table produced by TQCENL
Output of next call to TQCEL will be written to file 'result'
Output of subsequent calls to TQCEL/TQSHOW/TQMAPL will go to UNIT 6 again
! Get and display results ! First, we get the fraction of the first phase constituent of the first ! phase. We get also its name with TQGNPC. Since the current amount ! unit is 'mol', we get the 'mole fraction' (as opposed to 'mass ! fraction') CALL TQGNSC(1, DSTR, NOERR) IF (NOERR .NE. 0) CALL CABORT(739,'TQGNSC',NOERR) CALL TQGETR('xp ', 1, 1, D1, NOERR) IF (NOERR .NE. 0) CALL CABORT(741,'TQGETR',NOERR) WRITE(UNIT=*,FMT=51) 'Mole fraction of system component '//DSTR WRITE(UNIT=*,FMT=57) 'in the GAS phase: ', D1
Mole fraction of system component C in the GAS phase: 0.74334E-01
! Now get the equilibrium amount of the same constituent CALL TQGETR('a ',1,1,D1,NOERR) IF (NOERR .NE. 0) CALL CABORT(748,'TQGETR',NOERR) WRITE(UNIT=*,FMT=51) 'Equilibrium amount of '//DSTR WRITE(UNIT=*,FMT=57) 'in the GAS phase: ', D1
Equilibrium amount of C in the GAS phase: 0.0000
! Note that the result is zero, because the gas phase is not ! considered stable. The reason is that we are using the default ! ambient pressure of 1 bar for the calculations, and from the ! result table produced with the previous call to TQCEL or the ! subsequent call to TQGETR we see that the activity of the gas ! phase is less than unity, which means it is not considered ! stable, and thus the equilibrium amounts of all its phase ! constituents are zero. ! Using the option 'ac', TQGETR retrieves activities, in this case the ! activity (fugacity) of the gas phase CALL TQGETR('ac ',1, 0, D1, NOERR) IF (NOERR .NE. 0) CALL CABORT(765,'TQGETR',NOERR) WRITE(UNIT=*,FMT=58) 'Activity of the GAS phase: ', D1 ! Now TQGETR is used in a way that causes it to return an array. In ! this case, we are going to retrieve an array that contains the ! fugacities of all constituents of the gas phase. ! Get the fugacities: CALL TQGETR('ac ', 1, -1, DARR30, NOERR) IF (NOERR .NE. 0) CALL CABORT(775,'TQGETR',NOERR) ! Print them out, together with the names of the constituents ! (note that these values correspond to the last column of the ! result table previously output with TQCEL): WRITE(UNIT=*,FMT=51) 'Fugacities of all gas phase constituents:' DO I=1, NPCGAS CALL TQGNPC(1, I, DSTR, NOERR) IF (NOERR .NE. 0) CALL CABORT(785,'TQGNPC',NOERR) WRITE(UNIT=*,FMT=57) DSTR // ': ', DARR30(I) ENDDO WRITE(UNIT=*,FMT=51) ' '
Activity of the GAS phase: 0.14450 Fugacities of all gas phase constituents: C : 0.10955E-12 C2 : 0.19220E-15 C3 : 0.10532E-15 CO : 0.21481E-01 CO2 : 0.54503E-06 O : 0.42711E-11 O2 : 0.20213E-15 O3 : 0.96770E-31 Si : 0.12142E-04 Si2 : 0.10817E-06 Si2C : 0.20832E-06 Si3 : 0.23173E-07 SiC : 0.53182E-11 SiO : 0.12301 SiO2 : 0.39975E-07
! Get thermodynamic data of a phase constituent CALL TQGDPC('G ', 1, 1, D1, NOERR) IF (NOERR .NE. 0) CALL CABORT(796,'TQGDPC',NOERR) WRITE(UNIT=*,FMT=58) 'The (dimensionless) value of G for ' // * 'constituent 1 of phase 1 is: ', D1
The (dimensionless) value of G for constituent 1 of phase 1 is: 23.826
! Note that when retrieving CP, H, S, or G using TQGDPC, the values are ! returned 'dimensionless', which means they might have to be multiplied ! by R*T. Since the default amount unit is mol, results are returned for ! 1 mol. Note also that care has to be taken if a temperature unit ! different from Kelvin has been used. Refer to the manual entry of ! TQGDPC for a more detailed example. ! Check if we are working with the 'light' version. ! If we do, omit the following phase target calculation(s). CALL TQLITE(ISLITE, NOERR) IF (ISLITE .EQ. 1) THEN WRITE(*,FMT='(3(1X,A,/))') * '*** Phase target calculations have been omitted here,', * '*** since they are not possible with the', * '*** ''light'' version of ChemApp.' ELSE ! Perform a target calculation. This example demonstrates how to ! have ChemApp find the temperature at which a certain phase becomes ! stable. ! We want to have ChemApp calculate the temperature at which the ! phase 'SiO2(liquid)' becomes stable. First, determine the index ! number of this phase (note that it is allowed to abbreviate its ! name, as long as it is unambiguous): CALL TQINP('SiO2(liq', I, NOERR) IF (NOERR .NE. 0) CALL CABORT(828,'TQINP',NOERR) ! Next, define the formation target : CALL TQSETC('a ', I, 0, 0.D0, NUMCON, NOERR) IF (NOERR .NE. 0) CALL CABORT(832,'TQSETC',NOERR) ! Then call TQCEL to calculate the equilibrium, and pass it the ! information about the target variable (we want ChemApp to vary ! the _temperature_ until the phase is stable): WRITE(UNIT=*,FMT=51) 'Performing a phase target calculation' WRITE(UNIT=*,FMT=51) 'Phase target: formation of SiO2(liquid)' WRITE(UNIT=*,FMT=53) 'Target variable: Temperature' DARR2(1) = 2000.D0 WRITE(UNIT=*,FMT=51) '**** Begin output table produced by TQCEL' CALL TQCEL('t ',0,0,DARR2,NOERR) WRITE(UNIT=*,FMT=53) '**** End output table produced by TQCEL' IF (NOERR .NE. 0) CALL CABORT(846,'TQCEL',NOERR) DARR2(1) = 0.D0
Performing a phase target calculation
Phase target: formation of SiO2(liquid)
Target variable: Temperature
**** Begin output table produced by TQCEL
*T = 1995.99 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
CO/GAS/ 3.5701E-02
Si3/GAS/ 3.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
SiO 0.0000E+00 7.7442E-01 3.1442E-01
CO 0.0000E+00 2.2547E-01 9.1542E-02
Si 0.0000E+00 9.8748E-05 4.0093E-05
CO2 0.0000E+00 1.0313E-05 4.1873E-06
Si2C 0.0000E+00 2.5977E-06 1.0547E-06
Si2 0.0000E+00 1.1230E-06 4.5595E-07
SiO2 0.0000E+00 5.8253E-07 2.3651E-07
Si3 0.0000E+00 2.2928E-07 9.3089E-08
SiC 0.0000E+00 1.4260E-10 5.7898E-11
O 0.0000E+00 9.6352E-11 3.9120E-11
C 0.0000E+00 3.4767E-12 1.4116E-12
C2 0.0000E+00 1.2342E-14 5.0109E-15
O2 0.0000E+00 8.8186E-15 3.5804E-15
C3 0.0000E+00 8.5734E-15 3.4809E-15
O3 0.0000E+00 2.7760E-29 1.1271E-29
TOTAL: 0.0000E+00 1.0000E+00 4.0601E-01
mol ACTIVITY
Si 8.9464E+00 1.0000E+00
SiC 3.5701E-02 1.0000E+00
SiO2(cristobali) 1.7851E-02 1.0000E+00
SiO2(liquid) 0.0000E+00 1.0000E+00
SiO2(tridymite) T 0.0000E+00 9.9861E-01
SiO2(quartz) T 0.0000E+00 8.9882E-01
C 0.0000E+00 5.0396E-02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
2.46559E+02 8.32897E+05 8.66988E+02 -8.97600E+05 0.00000E+00
Data on 2 constituents marked with 'T' are extrapolated outside their valid
temperature range
**** End output table produced by TQCEL
! Note that in the resulting ChemSage table, the temperature is marked ! with an asterisk, meaning that it has been calculated by ChemApp, and ! the activity of SiO2(liquid) is unity, whereas its equilibrium amount ! is zero, meaning it just forms at this temperature ! Retrieve the calculated temperature: CALL TQGETR('t ', 0, 0, D1, NOERR) IF (NOERR .NE. 0) CALL CABORT(857,'TQGETR',NOERR) WRITE(UNIT=*,FMT=58) 'Calculated formation temperature of '// * 'SiO2(liquid): ', D1
Calculated formation temperature of SiO2(liquid): 1996.0
! One-dimensional phase mapping calculations. The example below ! demonstrates how to locate all phase transitions in a given ! temperature range. ! One-dimensional phase mapping calculations are only possible with ! version 3.x and later of ChemApp. WRITE(UNIT=*,FMT=51) 'Performing one-dimensional phase' // * ' mapping calculations' ! Initialise the variable to zero that holds the number of times TQMAP ! found results RESNO = 0 ! Remove all previous conditions CALL TQREMC(-2, NOERR) IF (NOERR .NE. 0) CALL CABORT(878,'TQREMC',NOERR) ! Determine the index number for the phase SiO2(quartz) CALL TQINP('SiO2(quartz) ', I, NOERR) IF (NOERR .NE. 0) CALL CABORT(882,'TQINP',NOERR) ! Enter one mol of SiO2 CALL TQSETC('IA ', I, 0, 1.0D0, NUMCON, NOERR) IF (NOERR .NE. 0) CALL CABORT(886,'TQSETC',NOERR) ! The temperature search interval is supposed to range from 300 to ! 3000 K: DARR2(1) = 300.D0 DARR2(2) = 3000.D0 ! First call to TQMAP, note the 'F' ('First') in the option parameter CALL TQMAP('TF ',0,0,DARR2,ICONT,NOERR) IF (NOERR .NE. 0) CALL CABORT(895,'TQMAP',NOERR) ! The variable RESNO keeps track of the number of times we called TQMAP: RESNO = RESNO + 1 ! Retrieve and print the temperature, which we know is DARR2(1) CALL TQGETR('T ',0,0,D1,NOERR) IF (NOERR .NE. 0) CALL CABORT(902,'TQGETR',NOERR) WRITE(*,59) '*** Lower interval boundary:', D1, ' K' 30 CONTINUE ! TQMAP is called again. Note the 'N' ('Next') in the option ! parameter. If we are at the first phase boundary (RESNO is still 2), ! we call TQMAPL for a change to produce a ChemSage output table... IF (RESNO .EQ. 2) THEN WRITE(*,FMT=51) * '*** ChemSage result table for the first phase ' // * 'boundary found:' WRITE(UNIT=*,FMT=51) * '**** Begin output table produced by TQMAPL' CALL TQMAPL('TN ',0,0,DARR2,ICONT,NOERR) IF (NOERR .NE. 0) CALL CABORT(919,'TQMAPL',NOERR) WRITE(UNIT=*,FMT=51) * '**** End output table produced by TQMAPL' IF (NOERR .NE. 0) CALL CABORT(922,'TQMAPL',NOERR) WRITE(*,*) ! ...otherwise we just call TQMAP: ELSE CALL TQMAP('TN ',0,0,DARR2,ICONT,NOERR) IF (NOERR .NE. 0) CALL CABORT(927,'TQMAP',NOERR) ENDIF RESNO = RESNO +1 ! Get the temperature... CALL TQGETR('T ',0,0,D1,NOERR) IF (NOERR .NE. 0) CALL CABORT(933,'TQGETR',NOERR) ! ... and print the entry for the result table. If we have called TQMAP ! twice already, we know that we found a phase boundary. If not, we have ! retrieved the temperature value of the upper interval boundary ! DARR2(2): IF (RESNO .GT. 2) THEN WRITE(*,59) '*** Phase boundary found at ', D1, ' K' ELSE WRITE(*,59) '*** Upper interval boundary: ', D1, ' K' ENDIF ! For as long as ICONT is positive, we need to make further calls to ! TQMAP IF (ICONT .GT. 0) GOTO 30
Performing one-dimensional phase mapping calculations
*** Lower interval boundary: 300.00 K
*** Upper interval boundary: 3000.00 K
*** ChemSage result table for the first phase boundary found:
**** Begin output table produced by TQMAPL
*T = 1140.10 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
SiO2(quartz) 1.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
SiO 0.0000E+00 6.2502E-01 4.4829E-16
O2 0.0000E+00 2.5088E-01 1.7994E-16
O 0.0000E+00 1.2327E-01 8.8415E-17
SiO2 0.0000E+00 8.3208E-04 5.9681E-19
Si 0.0000E+00 4.4651E-15 3.2026E-30
O3 0.0000E+00 2.4636E-19 1.7670E-34
Si2 0.0000E+00 5.5659E-36 3.9921E-51
Si3 0.0000E+00 9.0161E-54 6.4667E-69
TOTAL: 0.0000E+00 1.0000E+00 7.1725E-16
mol ACTIVITY
SiO2(quartz) 1.0000E+00 1.0000E+00
SiO2(tridymite) 0.0000E+00 1.0000E+00
SiO2(cristobali) 0.0000E+00 9.9398E-01
SiO2(liquid) 0.0000E+00 6.4439E-01
Si 0.0000E+00 2.5802E-17
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
7.04990E+01 -8.55398E+05 1.25413E+02 -9.98382E+05 0.00000E+00
**** End output table produced by TQMAPL
*** Phase boundary found at 1140.10 K
*** Phase boundary found at 1738.28 K
*** Phase boundary found at 1995.99 K
! With the above example the temperatures of all phase boundaries in a ! system which contains 1 mol of SiO2 have been calculated. Thus the ! phase boundaries determined reflect the stability ranges of the ! various modifications of SiO2. Also note again that the first two ! temperatures determined are _no_ phase boundaries, but the lower and ! upper limit of the search interval (DARR2(1) and DARR2(2)). ENDIF ! Since from now on we will be using streams instead of global ! conditions, we first remove all conditions and targets set up to this ! point. Note that using a '-1' instead of the '-2' in the call to ! TQREMC would also reset ChemApp to default units and values. CALL TQREMC(-2, NOERR) IF (NOERR .NE. 0) CALL CABORT(964,'TQREMC',NOERR) WRITE(UNIT=*,FMT=53) 'Calculations using streams:' ! Set name and temperature for a stream via the array TP, which ! will be passed to TQSTTP when each of the streams is created. TP(1) = 1000.D0 TP(2) = 1.D0 ! Create 3 streams: CALL TQSTTP('stream1 ', TP, NOERR) IF (NOERR .NE. 0) CALL CABORT(975,'TQSTTP',NOERR) CALL TQSTTP('stream2 ', TP, NOERR) IF (NOERR .NE. 0) CALL CABORT(977,'TQSTTP',NOERR) CALL TQSTTP('stream3 ', TP, NOERR) IF (NOERR .NE. 0) CALL CABORT(979,'TQSTTP',NOERR) ! Set constituent amounts for each stream CALL TQSTCA('stream1 ', 1, 4, 1.D0,NOERR) IF (NOERR .NE. 0) CALL CABORT(983,'TQSTCA',NOERR) CALL TQSTCA('stream2 ', 1, 12, 3.D0,NOERR) IF (NOERR .NE. 0) CALL CABORT(985,'TQSTCA',NOERR) CALL TQSTCA('stream3 ', 1, 8, 2.D0,NOERR) IF (NOERR .NE. 0) CALL CABORT(987,'TQSTCA',NOERR) ! Remove the last stream CALL TQSTRM('stream3 ', NOERR) IF (NOERR .NE. 0) CALL CABORT(991,'TQSTRM',NOERR) ! Set equilibrium temperature CALL TQSTEC('t ', 0, 1800.D0, NOERR) IF (NOERR .NE. 0) CALL CABORT(995,'TQSTEC',NOERR) ! Calculate and list equilibrium WRITE(UNIT=*,FMT=51) '**** Begin output table produced by TQCEL' CALL TQCEL(' ',0,0,DARR2,NOERR) WRITE(UNIT=*,FMT=53) '**** End output table produced by TQCEL' IF (NOERR .NE. 0) CALL CABORT(1001,'TQCEL',NOERR)
Calculations using streams:
**** Begin output table produced by TQCEL
T = 1800 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/K PRESSURE/bar STREAM
CO/GAS/ 1.0000E+00 1000.00 1.0000E+00 2
Si3/GAS/ 3.0000E+00 1000.00 1.0000E+00 3
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
SiO 0.0000E+00 9.1172E-01 4.1405E-02
CO 0.0000E+00 8.8214E-02 4.0062E-03
Si 0.0000E+00 6.7137E-05 3.0490E-06
CO2 0.0000E+00 1.1335E-06 5.1478E-08
Si2C 0.0000E+00 7.0003E-07 3.1792E-08
Si2 0.0000E+00 4.5031E-07 2.0451E-08
SiO2 0.0000E+00 1.1236E-07 5.1026E-09
Si3 0.0000E+00 1.0170E-07 4.6188E-09
SiC 0.0000E+00 7.3913E-12 3.3567E-13
O 0.0000E+00 7.2737E-12 3.3033E-13
C 0.0000E+00 1.2567E-13 5.7072E-15
O2 0.0000E+00 1.6031E-16 7.2803E-18
C2 0.0000E+00 9.7381E-17 4.4225E-18
C3 0.0000E+00 4.0495E-17 1.8391E-18
O3 0.0000E+00 8.7034E-33 3.9526E-34
TOTAL: 0.0000E+00 1.0000E+00 4.5415E-02
mol ACTIVITY
Si 7.5000E+00 1.0000E+00
SiC 1.0000E+00 1.0000E+00
SiO2(cristobali) 5.0000E-01 1.0000E+00
SiO2(tridymite) 0.0000E+00 9.9970E-01
SiO2(liquid) 0.0000E+00 9.3907E-01
SiO2(quartz) T 0.0000E+00 9.1491E-01
C 0.0000E+00 2.2526E-02
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
7.76667E+01 -1.68039E+06 -3.77532E+02 -2.00297E+06 -3.32581E+02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
2.93963E+02 2.64115E+05 8.75143E+02 -1.31114E+06 0.00000E+00
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
**** End output table produced by TQCEL
! Get thermodynamic properties of a stream (note that as of version ! 3.2.0 of ChemApp, TQSTXP can also be called _before_ the equilibrium ! calculation) CALL TQSTXP('stream1 ', 'H ', D1, NOERR) IF (NOERR .NE. 0) CALL CABORT(1009,'TQSTXP',NOERR) WRITE(UNIT=*,FMT=58) 'Enthalpy of ''stream1'': ', D1
Enthalpy of 'stream1': -88842.
WRITE(UNIT=*, FMT=53) 'Demonstrating the use of ChemApp ' //
* 'subroutines involving sublattices.'
! To demonstrate the subroutines dealing with sublattices, a different
! data-file (subl-ex.dat) needs to be loaded. This data-file contains an
! extract of the system Co-Cr-Fe: the phase SIGMA:30, which is modelled
! according to the sublattice formalism, and the BCC phase, described by
! a Redlich-Kister polynomial. Both phases are each included twice, to
! account for miscibility gaps.
CALL TQOPNA('subl-ex.dat', UNITNO, NOERR)
IF (NOERR .NE. 0) CALL CABORT(1025,'TQOPNA',NOERR)
! Read data-file
CALL TQRFIL(NOERR)
IF (NOERR .NE. 0) CALL CABORT(1029,'TQRFIL',NOERR)
! Close data-file
CALL TQCLOS(UNITNO, NOERR)
IF (NOERR .NE. 0) CALL CABORT(1033,'TQCLOS',NOERR)
! The first of the two identical copies of the SIGMA:30 phase, which
! ChemApp calls SIGMA:30#1, will be investigated with respect to the
! number of sublattices, the number of sublattice constituents on each
! sublattice, and the names of the sublattice constituents.
! Get the index number for the phase SIGMA:30#1
PNAME = 'SIGMA:30#1'
CALL TQINP(PNAME, INDEXP, NOERR)
IF (NOERR .NE. 0) CALL CABORT(1043,'TQINP',NOERR)
! Get the number of sublattices
CALL TQNOSL(INDEXP, NSL, NOERR)
IF (NOERR .NE. 0) CALL CABORT(1047,'TQNOSL',NOERR)
WRITE(UNIT=*,FMT=60) PNAME // ' has ', NSL, ' sublattices'
Demonstrating the use of ChemApp subroutines involving sublattices. SIGMA:30#1 has 3 sublattices
! Loop over all sublattices DO INDEXL=1, NSL ! Get the number of sublattice constituents CALL TQNOLC(INDEXP, INDEXL, NSLCON, NOERR) IF (NOERR .NE. 0) CALL CABORT(1056,'TQNOLC',NOERR) WRITE(UNIT=*,FMT=56) 'Sublattice ', INDEXL, ' has ', NSLCON, * ' constituents with the following names:' ! Get the name of each sublattice constituent DO INDEXC=1, NSLCON CALL TQGNLC(INDEXP, INDEXL ,INDEXC, CNAME, NOERR) IF (NOERR .NE. 0) CALL CABORT(1063,'TQGNLC',NOERR) ! The reverse (getting the index number for the name of the sublattice ! constituent just retrieved), is rather superfluous here, and only used ! to demonstrate the call to TQINLC: CALL TQINLC(CNAME, INDEXP, INDEXL, I, NOERR) IF (NOERR .NE. 0) CALL CABORT(1069,'TQINLC',NOERR) WRITE(UNIT=*,FMT=60) ' ',I,':' // CNAME ENDDO ENDDO
Sublattice 1 has 2 constituents with the following names:
1:Co
2:Fe
Sublattice 2 has 1 constituents with the following names:
1:Cr
Sublattice 3 has 3 constituents with the following names:
1:Co
2:Cr
3:Fe
! Set the temperature to 1000 K CALL TQSETC('T ', 0, 0, 1000.D0, NUMCON, NOERR) IF (NOERR .NE. 0) CALL CABORT(1078,'TQSETC',NOERR) ! Set the incoming amounts to 0.25 mol Co, 0.25 mol Cr, and 0.5 mol Fe CALL TQINSC('Co ', INDEXC, NOERR) IF (NOERR .NE. 0) CALL CABORT(1082,'TQINSC',NOERR) CALL TQSETC('IA ', 0, INDEXC, 0.25D0, NUMCON, NOERR) IF (NOERR .NE. 0) CALL CABORT(1084,'TQSETC',NOERR) CALL TQINSC('Cr ', INDEXC, NOERR) IF (NOERR .NE. 0) CALL CABORT(1087,'TQINSC',NOERR) CALL TQSETC('IA ', 0, INDEXC, 0.25D0, NUMCON, NOERR) IF (NOERR .NE. 0) CALL CABORT(1089,'TQSETC',NOERR) CALL TQINSC('Fe ', INDEXC, NOERR) IF (NOERR .NE. 0) CALL CABORT(1092,'TQINSC',NOERR) CALL TQSETC('IA ', 0, INDEXC, 0.50D0, NUMCON, NOERR) IF (NOERR .NE. 0) CALL CABORT(1094,'TQSETC',NOERR) WRITE(UNIT=*, FMT=55) 'Calculate the equilibrium, ' // * 'get information on the stable sublattice phases.' ! Calculate the equilibrium DARR2(1) = 0.D0 CALL TQCE(' ', 0, 0, DARR2, NOERR) IF (NOERR .NE. 0) CALL CABORT(1103,'TQCE',NOERR) ! Get information on the stable sublattice phases. ! Loop over all phases, and if a phase is stable and a sublattice phase, ! print information about the sublattices CALL TQNOP(NPHASE, NOERR) IF (NOERR .NE. 0) CALL CABORT(1110,'TQNOP',NOERR) DO INDEXP=1, NPHASE ! Check if the phase is stable, otherwise we don't need to consider it CALL TQGETR('A ', INDEXP, 0, D1, NOERR) IF (NOERR .NE. 0) CALL CABORT(1116,'TQGETR',NOERR) IF (D1 .GT. 0.D0) THEN ! Get the name and model of the phase CALL TQGNP(INDEXP, PNAME, NOERR) IF (NOERR .NE. 0) CALL CABORT(1121,'TQGNP',NOERR) CALL TQMODL(INDEXP, MNAME, NOERR) IF (NOERR .NE. 0) CALL CABORT(1123,'TQMODL',NOERR) ! If the model name of the phase starts with 'SUB' we have a sublattice ! phase IF (INDEX(MNAME,'SUB') .EQ. 1) THEN ! Print a header similar to the one in the ChemSage output table WRITE(UNIT=*,FMT=51) 'Mole fraction of the ' // * 'sublattice constituents in ' // PNAME ! Get the number of sublattices CALL TQNOSL(INDEXP, NSL, NOERR) IF (NOERR .NE. 0) CALL CABORT(1135,'TQNOSL',NOERR) ! Loop over all sublattices DO INDEXL=1, NSL write(UNIT=*,FMT=60) 'Sublattice ',INDEXL,' :' ! Get the number of sublattice constituents CALL TQNOLC(INDEXP, INDEXL, NSLCON, NOERR) IF (NOERR .NE. 0) CALL CABORT(1143,'TQNOLC', * NOERR) DO INDEXC=1, NSLCON ! Get the name of each sublattice constituent... CALL TQGNLC(INDEXP, INDEXL, INDEXC, CNAME, NOERR) IF (NOERR .NE. 0) CALL CABORT(1149,'TQGNLC', * NOERR) ! ... and its mole fraction in the sublattice CALL TQGTLC(INDEXP, INDEXL, INDEXC, D1, NOERR) IF (NOERR .NE. 0) CALL CABORT(1154,'TQGTLC', * NOERR) WRITE(UNIT=*,FMT=57) CNAME, D1 ENDDO ENDDO ENDIF ENDIF ENDDO WRITE(UNIT=*,FMT=51) ' '
Calculate the equilibrium, get information on the stable sublattice phases. Mole fraction of the sublattice constituents in SIGMA:30#1 Sublattice 1 : Co 0.54238 Fe 0.45762 Sublattice 2 : Cr 1.0000 Sublattice 3 : Co 0.10926 Cr 0.69192 Fe 0.19882
! The following section demonstrates the use of subroutines that were ! introduced in ChemApp V4.0.0 to support 'transparent data-files'. ! Retrieve the licensee's user ID CALL TQGTID(ID, NOERR) IF (NOERR .NE. 0) CALL CABORT(1173,'TQGTID',NOERR) ! Retrieve the licensee's name CALL TQGTNM(NAME, NOERR) IF (NOERR .NE. 0) CALL CABORT(1177,'TQGTNM',NOERR) ! Retrieve the program ID CALL TQGTPI(PID, NOERR) IF (NOERR .NE. 0) CALL CABORT(1181,'TQGTPI',NOERR) ! Print all three WRITE(UNIT=*,FMT=51) 'Licensee''s user ID: ' // * ID(1:STRLEN(ID)) WRITE(UNIT=*,FMT=51) 'Licensee''s name : ' // * NAME(1:STRLEN(NAME)) WRITE(UNIT=*,FMT=53) 'Program ID : ' // * PID(1:STRLEN(PID))
Licensee's user ID: 5001 Licensee's name : GTT - Technologies Program ID : CAFU
! Get the ChemApp license expiration date (month and year) CALL TQGTED(EDMON, EDYEAR, NOERR) IF (NOERR .NE. 0) CALL CABORT(1194,'TQGTED',NOERR) WRITE(UNIT=*,FMT=62) 'ChemApp license expiration date ' // * '(month/year): ', EDMON, '/', EDYEAR
ChemApp license expiration date (month/year): 12/2025
! The following pieces of information are only meaningful if a version ! of ChemApp is used that requires a dongle (hardware key). ! Get the HASP dongle type and id CALL TQGTHI(HASPT, HASPID, NOERR) IF (NOERR .NE. 0) CALL CABORT(1204,'TQGTHI',NOERR) ! Print info if HASP dongle is used: IF (HASPID .NE. 0) THEN WRITE(UNIT=*,FMT=51) 'HASP dongle type : ' // * HASPT(1:STRLEN(HASPT)) WRITE(UNIT=*,FMT=63) 'HASP dongle id : ', HASPID ELSE WRITE(UNIT=*,FMT=53) 'This ChemApp version does not ' // * 'require a HASP hardware key (dongle)' ENDIF
This ChemApp version does not require a HASP hardware key (dongle)
! Get the default unit number associated with error output CALL TQGIO('ERROR ', EUNIT, NOERR) IF (NOERR .NE. 0) CALL CABORT(1219,'TQGIO',NOERR) ! Turn off automatic error reporting for the next call to TQCIO. CALL TQCIO('ERROR ', 0, NOERR) IF (NOERR .NE. 0) CALL CABORT(1223,'TQCIO',NOERR) ! The following section is only executed if the file "cosiex.cst", ! a sample thermochemical data-file in transparent format, is ! present. It is not included in regular distributions of ChemApp CALL TQOPNT('cosiex.cst', UNITNO, I) ! Turn automatic error reporting back on. CALL TQCIO('ERROR ', EUNIT, NOERR) IF (NOERR .NE. 0) CALL CABORT(1232,'TQCIO',NOERR) IF (I .NE. 0) THEN WRITE(UNIT=*,FMT=*) 'Skipping transparent file ' // * 'subroutines, since file "cosiex.cst" cannot be read' ELSE ! Read data-file CALL TQRCST(NOERR) IF (NOERR .NE. 0) CALL CABORT(1241,'TQRCST',NOERR) ! Close data-file CALL TQCLOS(UNITNO, NOERR) IF (NOERR .NE. 0) CALL CABORT(1245,'TQCLOS',NOERR) ! Once the transparent data-file has been read, information on its ! header can be retrieved. CALL TQGTRH(TFHVER, TFHNWP, TFHVNW, TFHNRP, TFHVNR, TFHDTC, * TFHDTE, TFHID, TFHUSR, TFHREM, NOERR) IF (NOERR .NE. 0) CALL CABORT(1251,'TQGTRH',NOERR) WRITE(UNIT=*, FMT=*) 'Version number of the transparent ' // * 'file header format:', TFHVER WRITE(UNIT=*, FMT=*) 'Name of the program which wrote the ' // * 'data-file: ', TFHNWP(1:STRLEN(TFHNWP)) WRITE(UNIT=*, FMT=*) 'Version number of the writing program: ', * TFHVNW(1), '.', TFHVNW(2), '.', TFHVNW(3) WRITE(UNIT=*, FMT=*) 'Programs which are permitted to read ' // * 'the data-file: ', TFHNRP(1:STRLEN(TFHNRP)) WRITE(UNIT=*, FMT=*) 'Min. version number of the reading ' // * 'program: ', TFHVNR(1), '.', TFHVNR(2), '.', TFHVNR(3) WRITE(UNIT=*, FMT=61) 'File was created on ', * TFHDTC(1),'/', TFHDTC(2),'/',TFHDTC(3),' ', * TFHDTC(4),':',TFHDTC(5),':',TFHDTC(6) WRITE(UNIT=*, FMT=61) 'File will expire on ', * TFHDTE(1),'/', TFHDTE(2),'/',TFHDTE(3),' ', * TFHDTE(4),':',TFHDTE(5),':',TFHDTE(6) WRITE(UNIT=*, FMT=*) 'Licensee''s user ID(s): ', * TFHID(1:STRLEN(TFHID)) WRITE(UNIT=*, FMT=*) 'Licensee''s name: ', * TFHUSR(1:STRLEN(TFHUSR)) WRITE(UNIT=*, FMT=*) 'Remarks: ', * TFHREM(1:STRLEN(TFHREM))
Version number of the transparent file header format: 1 Name of the program which wrote the data-file: FactSage Version number of the writing program: 5 . 0 . 0 Programs which are permitted to read the data-file: CAFU,CALI Min. version number of the reading program: -1 . -1 . -1 File was created on 2015/09/09 15:13:52 File will expire on 2036/12/31 12:00:00 Licensee's user ID(s): ???? Licensee's name: GTT - Technologies Remarks: For use with ChemApp example programs
ENDIF WRITE(UNIT=*,FMT=55) 'End of output from cademo1.' END
End of output from cademo1.
13 C example code (cademo1.c)
For explanatory notes on this example program, please refer to FORTRAN example code (cademo1.f).
/* Note that this program contains phase target calculations, which cannot be performed with the 'light' version of ChemApp */ /* Please also read the notes regarding the use of ChemApp with C/C++, which are contained in a separate file. Please direct any questions about this program or the results it produces on your machine to: GTT-Technologies Kaiserstrasse 100 52134 Herzogenrath Germany Phone: +49-2407-59533 Fax: +49-2407-59661 E-mail: support@gtt-technologies.de WWW: http://www.gtt-technologies.de/ */ #include <stdio.h> #include <stdlib.h> #include <string.h> #include "cacint.h" /* --------------------------------------------------------------------- * Func : abortprog * --------------------------------------------------------------------- * Subject : If a ChemApp error occurs, this function reports the error number and the routine it occurred in before it exits the program. * * --------------------------------------------------------------------- */ void abortprog(int lineno, char sr_name[10], LI error_no) { fprintf(stdout,"\nChemApp error no. %li occurred when calling %s.\n" "Aborting on line %i of %s .\n", error_no, sr_name, lineno, __FILE__); exit(error_no); } /* --------------------------------------------------------------------- * Func : main * --------------------------------------------------------------------- * Subject : Main body of program * * --------------------------------------------------------------------- */ int main() { /* Declaration of variables to be used in the program */ /* Every integer variable that is to be used in an invocation of a ChemApp subroutine has to be of type "LI". "LI" is defined in cacint.h and is usually an abbreviation for "long int". Similarly, every real variable for use with ChemApp subroutines has to be of type "DB", which is usually an abbreviation for "double" */ /* Declarations of integer variables for use with ChemApp: */ /* An all-purpose integer variable */ LI lint, /* Integer variables which hold the index number of phases, phase constituents, and sublattices */ indexp, indexc, indexl, /* The standard error code return variable */ noerr, /* A variable that is set to the version number of ChemApp */ cavers, /* This variable is used in conjunction with tqlite to determine whether the program is linked to the regular or the 'light' version of ChemApp. In the latter case, target calculations are left out. */ islite, /* A number of integer variables to return the information from tqsize */ lia,lib,lic,lid,lie,lif,lig,lih,lii,lij,lik, /* A similar set of integer variables to return the information from tqused */ ldia,ldib,ldic,ldid,ldie,ldif,ldig,ldih,ldii,ldij,ldik, /* An integer that will contain the index number of a condition set */ numcon, /* An integer that will contain the number of phases in the data-file loaded */ nphase, /* An integer that will hold the number of phase constituents in the gas phase */ npcgas, /* An integer that will contain the number of system components */ nscom, /* Integers that will contain the number of sublattices, and the number of sublattice contituents of a particular sublattice */ nsl, nslcon, /* Two integers used for one-dimensional phase mapping calculations, one to indicate whether we need to make more calls to tqmap/tqmapl */ icont, /* And another one to keep track of the number of results we obtained */ resno, /* An integer that holds the FORTRAN unit number under which the thermochemical data-files will be opened */ unitno, /* An integer that holds the FORTRAN unit number to which the ChemApp error messages are written by default */ errorunit, /* Variables to store information from various transparent file header fields */ tfhver, tfhvnw[3], tfhvnr[3], tfhdtc[6], tfhdte[6], /* These integers will be used to store the HASP dongle id, plus the ChemApp expiration month and year */ haspid, edmon, edyear; /* Declarations of real variables for use with ChemApp: */ /* An all-purpose real variable */ DB d1, /* An array of two real variables... */ darray2[2], /* ...and one of 30 */ darray30[30], /* ...and one for which memory will be allocated dynamically to be just of the right size */ *darray, /* A variable to hold the molecular mass of a system component (see tqstsc) or a phase constituent (see tqstpc) */ wmass, /* This particular array of two real variables will contain the temperature and pressure of a stream to be defined with tqsttp */ tp[2]; /* Declarations of string variables for use with ChemApp: */ /* Every "name" used in conjuction with ChemApp, whether it is the name of a phase, a system component, a phase constituent, or a stream, can be up to 24 characters in length (Exceptions are the subroutine tqerr, as well as the subroutines tqgtrh, tqgtid, tqgtnm, tqgtpi, and tqwstr, which all pass or return strings longer than 24 characters). This means that in C/C++, these strings have to be 25 characters long, due to the trailing null character (\0). TQSTRLEN is defined in cacint.h to be 25. Thus, if you define all your strings to be used with ChemApp using TQSTRLEN, you can be sure that they will have the right length. Another way to do this is to use the macro "TQSTRING(x)", as defined in cacint.h, which expands to "char x[TQSTRLEN]". */ /* an all-purpose static string for ChemApp */ char dstr[TQSTRLEN], /* A pointer to a string, before it can be used, sufficient memory has to be allocated */ *dstrptr, /* Character strings that will hold names of phases, phase constituents, system components, mixture model names, as well as sublattatices and their constituents */ pname[TQSTRLEN], cname[TQSTRLEN], mname[TQSTRLEN], /* The following variable is an array of 4 strings. It will be used with tqcsc, which is the only ChemApp subroutine that takes a character array as input. Please note that the last string element of such an array for use with tqcsc must be an extra empty string, this is due to the peculiarities of exchanging strings between FORTRAN and C code. This is why we declare the following array to have 4 elements, although we will only have 3 system components. */ newsc[4][TQSTRLEN]; /* Using the macro TQSTRING to declare a string variable for ChemApp*/ TQSTRING(dstr2); /* The next three character strings are used to hold information about the copy of ChemApp used. */ char id[255], name[80], pid[TQSTRLEN]; /* Variables to store information from various transparent file header fields */ char tfhid[255], tfhusr[80],tfhnwp[40],tfhnrp[40],tfhrem[80]; /* This character variable will be used to store the HASP dongle type */ char haspt[TQSTRLEN]; /* Declaration of various other variables: */ int i; /* Print the program title and its ID */ printf("\nThis is cademo1, a C program to test ChemApp and its" " C Interface.\n\n");
This is cademo1, a C program to test ChemApp and its C Interface.
/* Allocate memory for the dynamic string variable dstrptr. By using TQSTRLEN, we make sure it has the right length. */ dstrptr = malloc(TQSTRLEN); /* Initialise ChemApp */ tqini(&noerr); /* After all following invocations of ChemApp subroutines, the error variable will be checked for a non-zero value, in which case an error has occurred and the program is aborted */ if (noerr) abortprog(__LINE__,"tqini",noerr); /* Print the copyright string. The way to do this is to first call the subroutine tqcprt, which copies the copyright string into the ChemApp-internal error buffer string: */ tqcprt(&noerr); if (noerr) abortprog(__LINE__,"tqcprt",noerr); /* Then do what is usually done to retrieve an error message: call tqerr to retrieve it. The variable TQERRMSG is declared in cacint.c as TQERRMSG[3][80] for this purpose:*/ tqerr((CHP)TQERRMSG,&noerr); /* Note that an explicit typecast has been used in the above call to tqerr to avoid compiler warnings about TQERRMSG being an incompatible pointer type ("CHP" is declared in cacint.h to be an abbreviation for "char*") */ if (noerr) abortprog(__LINE__,"tqerr",noerr); /* To print out the message, loop over the three strings of TQERRMSG: */ for(i=0;i<3;i++) printf("%s\n",TQERRMSG[i]);
This program contains ChemApp Copyright GTT-Technologies, Kaiserstrasse 100, D-52134 Herzogenrath, Germany http://www.gtt-technologies.de
/* Get version number of ChemApp*/ tqvers(&cavers,&noerr); if (noerr) abortprog(__LINE__,"tqvers",noerr); printf("\nChemApp version is: %i\n\n", cavers); /* Get array sizes */ tqsize(&lia,&lib,&lic,&lid,&lie,&lif,&lig,&lih,&lii,&lij,&lik,&noerr); if (noerr) abortprog(__LINE__,"tqsize",noerr); printf("Internal array sizes of this version of ChemApp,\n" "maximum number of:\n" "constituents: %li\n" "system components: %li\n" "mixture phases: %li\n" "excess Gibbs energy coefficients for " "a mixture phase: %li\n" "excess magnetic coefficients for " "a mixture phase: %li\n" "sublattices for a mixture phase: %li\n" "constituents of a sublattice: %li\n" "oxide constituents of a phase described by the " "Gaye-Kapoor-Frohberg\n" " or modified quasichemical formalisms: %li\n" "Gibbs energy/heat capacity equations for " "a constituent: %li\n" "Gibbs energy/heat capacity equations: %li\n" "constituents with P,T-dependent " "molar volumes: %li\n\n", lia,lib,lic,lid,lie,lif,lig,lih,lii,lij,lik);
ChemApp version is: 645 Internal array sizes of this version of ChemApp, maximum number of: constituents: 1600 system components: 48 mixture phases: 44 excess Gibbs energy coefficients for a mixture phase: 1999 excess magnetic coefficients for a mixture phase: 499 sublattices for a mixture phase: 140724603453445 constituents of a sublattice: 140724603455040 oxide constituents of a phase described by the Gaye-Kapoor-Frohberg or modified quasichemical formalisms: 140724603453465 Gibbs energy/heat capacity equations for a constituent: 140724603453460 Gibbs energy/heat capacity equations: 140724603458240 constituents with P,T-dependent molar volumes: 140724603455040
/* Get value for input/output option */ /* Determine which FORTRAN unit is used by default by tqrfil for reading the thermochemical data-file */ tqgio("FILE", &unitno, &noerr); if (noerr) abortprog(__LINE__,"tqgio",noerr); printf("The thermochemical data will be read from the file " "associated with unit %li\n\n", unitno);
The thermochemical data will be read from the file associated with unit 10
/* Open data-file for reading */ /* Note that tqopna is used to open the thermochemical data-file, instead of a standard C library routine like fopen. The reason is that it is necessary to associate the data-file with a particular FORTRAN unit number (10 by default, see above) so that the FORTRAN subroutine tqrfil can read from the correct file */ tqopna("cosi.dat",unitno,&noerr); if (noerr) abortprog(__LINE__,"tqopna",noerr); /* Read data-file */ tqrfil(&noerr); if (noerr) abortprog(__LINE__,"tqrfil",noerr); /* Close data-file */ /* Again, the routine for closing the data-file is not a standard C library routine like fclose, but a special one to make sure the file that was previously opened under the unit number specified is closed */ tqclos(unitno,&noerr); if (noerr) abortprog(__LINE__,"tqclos",noerr); /*C*# label(tqused) */ /* tqused is used to find out to what extent the thermochemical data storage space in the internal arrays of ChemApp (as reported by tqsize) are occupied by the currently loaded thermochemical system. */ tqused(&ldia,&ldib,&ldic,&ldid,&ldie,&ldif,&ldig,&ldih,&ldii, &ldij,&ldik,&noerr); if (noerr) abortprog(__LINE__,"tqused",noerr); /* As an example, check how many Gibbs energy/heat capacity equations are currently being used. */ printf("Actual number of Gibbs energy/heat capacity equations " "used (tqused): %li\n", ldij); printf("Maximum number available in this version of ChemApp " "(tqsize): %li\n", lij);
Actual number of Gibbs energy/heat capacity equations used (tqused): 35 Maximum number available in this version of ChemApp (tqsize): 4800
/* Get system units */ tqgsu("Pressure",dstr,&noerr); if (noerr) abortprog(__LINE__,"tqgsu",noerr); printf("Pressure unit: %s\n", dstr); tqgsu("Volume",dstr,&noerr); if (noerr) abortprog(__LINE__,"tqgsu",noerr); printf("Volume unit: %s\n", dstr); tqgsu("Temperature",dstr,&noerr); if (noerr) abortprog(__LINE__,"tqgsu",noerr); printf("Temperature unit: %s\n", dstr); tqgsu("Energy",dstr,&noerr); if (noerr) abortprog(__LINE__,"tqgsu",noerr); printf("Energy unit: %s\n", dstr); /* Change system unit */ /* Here we change the unit for the quantity "amount" to gram, mainly so that when we call tqstsc further down, we get the molecular mass expressed in the unit g/mol */ tqcsu("Amount","gram", &noerr); if (noerr) abortprog(__LINE__,"tqcsu",noerr); printf("Amount unit set to gram\n\n");
Pressure unit: bar Volume unit: dm3 Temperature unit: K Energy unit: J Amount unit set to gram
/* Get number of system components */ tqnosc(&nscom,&noerr); if (noerr) abortprog(__LINE__,"tqnosc",noerr); printf("Number of system components: %li\n\n", nscom); /* Get stoichiometry of system component */ /* Note that the array used (darray30) is much bigger than needed, which doesn't matter to ChemApp, as long as it is big enough. */ tqstsc(1,darray30,&wmass,&noerr); if (noerr) abortprog(__LINE__,"tqstsc",noerr); /* Here the array elements are printed, which describe the stoichiometry of the system component. This particular piece of information is not too useful though, since the array contains all zeros, except for a "1.0" at the position of the system component we specified */ printf("Stoichiometry of system component 1: "); for (i=1;i<=nscom;i++) printf("%f ", darray30[i-1]); /* Had we not changed the amount unit from the default value "mol" to "gram", we would receive a value of 1.0 for wmass. The reason is that the molecular mass is always output using the unit [current amount unit]/mol. If the current amount unit happens to be mol, the value for wmass will be 1.0 */ printf("\nMolecular mass in g/mol: %f\n", wmass); /* Get name of system component #1 */ tqgnsc(1,dstr,&noerr); if (noerr) abortprog(__LINE__,"tqgnsc",noerr); printf("Name of system component 1: %s\n", dstr); /* Get index number of system component */ /* This is the reverse of the above, using the name of the system component to get its index number */ tqinsc(dstr, &lint, &noerr); if (noerr) abortprog(__LINE__,"tqinsc",noerr); printf("Index number of system component %s is: %li\n\n",dstr,lint);
Number of system components: 3 Stoichiometry of system component 1: 1.000000 0.000000 0.000000 Molecular mass in g/mol: 12.011000 Name of system component 1: C Index number of system component C is: 1
/* Change system components */ /* tqcsc is the only ChemApp subroutine which takes an array of strings as input. The array contains as many strings as there are system components in the data-file, plus an extra one is needed when programming in C, which has to be empty. */ /* The system components are changed to SiO, SiC, and CO. */ strcpy(newsc[0], "SiO"); strcpy(newsc[1], "SiC"); strcpy(newsc[2], "CO"); /* Don't forget: The last string of the string array that will be passed to tqcsc has to be empty! */ strcpy(newsc[3], ""); tqcsc((CHP)newsc,&noerr); /* Note that an explicit typecast has been used in the above call to tqcsc to avoid compiler warnings about newsc being an incompatible pointer type */ if (noerr)abortprog(__LINE__,"tqcsc",noerr); printf("System components changed to SiO-SiC-CO.\n"); /* Print names of new system components */ for (lint = 1; lint <= nscom; lint++) { tqgnsc(lint,dstr,&noerr); if (noerr) abortprog(__LINE__,"tqgnsc",noerr); printf("Name of new system component %li: %s\n", lint, dstr); } /* Get stoichiometry of system component #1 */ tqstsc(1,darray30,&d1,&noerr); if (noerr) abortprog(__LINE__,"tqstsc",noerr); /* Note that the stoichiometry of the system component #1 does not change, as compared to the first call to tqstsc before the system components have been altered: */ printf("Stoichiometry of system component 1: "); for (i=1;i<=nscom;i++) printf("%f ", darray30[i-1]); /* What changes of course is the molecular mass: */ printf("\nMolecular mass in g/mol: %f\n\n", d1);
System components changed to SiO-SiC-CO. Name of new system component 1: SiO Name of new system component 2: SiC Name of new system component 3: CO Stoichiometry of system component 1: 1.000000 0.000000 0.000000 Molecular mass in g/mol: 44.085400
/* Get number of phases */ tqnop(&nphase,&noerr); if (noerr) abortprog(__LINE__,"tqnop",noerr); printf("Number of phases: %li\n", nphase); /* Get name of phase #1 */ tqgnp(1,dstr,&noerr); if (noerr) abortprog(__LINE__,"tqgnp",noerr); printf("Name of phase 1: %s\n", dstr); /* Get index number of phase */ /* This is the reverse of the above, using the name of the phase to get its index number */ tqinp(dstr,&lint,&noerr); if (noerr) abortprog(__LINE__,"tqinp",noerr); printf("Index number of phase %s is: %li\n",dstr,lint); /* Get model name of phase */ tqmodl(1, dstr, &noerr); if (noerr) abortprog(__LINE__,"tqmodl",noerr); printf("Model name of phase 1: %s\n",dstr); /* Whereas above we asked for the model name of the first phase in the data-file, which is normally a mixture phase (gas phase), we now ask for the model name of the last phase in the data-file (nphase, as determined by a call to tqnop). This is typically a stoichiometric condensed phase, in which case the string "PURE" is returned as the model name*/ tqmodl(nphase, dstr, &noerr); if (noerr) abortprog(__LINE__,"tqmodl",noerr); printf("Model name of phase %li: %s\n\n",nphase,dstr); /* Get number of phase constituents of phase #1 */ tqnopc(1, &npcgas, &noerr); if (noerr) abortprog(__LINE__,"tqnopc",noerr); printf("Number of phase constituents in phase 1: %li\n", npcgas); /* Get name of phase constituent */ tqgnpc(1, 1, dstr, &noerr); if (noerr) abortprog(__LINE__,"tqgnpc",noerr); printf("Name of phase constituent 1 of phase 1: %s\n", dstr); /* Get index number of phase constituent */ /* This is the reverse of the above, using the name of the phase constituent to get its index number */ tqinpc(dstr, 1, &lint, &noerr); if (noerr) abortprog(__LINE__,"tqinpc",noerr); printf("Index number of phase constituent %s is: %li\n",dstr,lint); /* Check whether the phase constituent can be used as incoming species (note that there are only few cases, and only for certain mixture phase models, where this might not be permitted)*/ tqpcis(1, 1, &lint, &noerr); if (noerr) abortprog(__LINE__,"tqpcis",noerr); if (lint == 0) printf("Usage of %s as incoming species is not permitted.\n", dstr); else printf("Usage of %s as incoming species is o.k.\n", dstr); /* Get stoichiometry of phase constituent */ /* tqstpc is a subroutine similar to tqstsc, except that with tqstpc, the returned stoichiometry array contains valuable information. This stoichiometry array represents the stoichiometry matrix for a phase constituent, as described in the ChemApp manual*/ tqstpc(1, 1, darray30, &wmass, &noerr); if (noerr) abortprog(__LINE__,"tqstpc",noerr); printf("Stoichiometry of phase constituent 1 in phase 1 is: "); for (i=1;i<=nscom;i++) printf("%f ", darray30[i-1]); printf("\nMolecular mass: %f\n\n", wmass);
Number of phases: 8 Name of phase 1: GAS Index number of phase GAS is: 1 Model name of phase 1: IDMX Model name of phase 8: PURE Number of phase constituents in phase 1: 15 Name of phase constituent 1 of phase 1: C Index number of phase constituent C is: 1 Usage of C as incoming species is o.k. Stoichiometry of phase constituent 1 in phase 1 is: -0.500000 0.500000 0.500000 Molecular mass: 12.011000
/* The system components are changed back to the original set of C, O, and Si, since we don't need the modified set of system components (SiO, SiC, and CO) any longer. */ strcpy(newsc[0], "C"); strcpy(newsc[1], "O"); strcpy(newsc[2], "Si"); strcpy(newsc[3], ""); tqcsc((CHP)newsc,&noerr); if (noerr)abortprog(__LINE__,"tqcsc",noerr); printf("System components changed back to C-O-Si.\n\n");
System components changed back to C-O-Si.
/* Changing the status of phases and phase constituents */ /* Change status of phase #1 */ tqcsp(1, "eliminated", &noerr); if (noerr) abortprog(__LINE__,"tqcsp",noerr); printf("Status of phase 1 set to 'eliminated'\n"); /* Get status of phase #1 */ tqgsp(1, dstr, &noerr); if (noerr) abortprog(__LINE__,"tqgsp",noerr); printf("Status of phase 1 is: %s \n", dstr); /* Change status of phase #1 */ tqcsp(1, "entered", &noerr); if (noerr) abortprog(__LINE__,"tqcsp",noerr); printf("Status of phase 1 set to 'entered'\n"); /* Get status of phase #1 */ tqgsp(1, dstr, &noerr); if (noerr) abortprog(__LINE__,"tqgsp",noerr); printf("Status of phase 1 is: %s \n", dstr);
Status of phase 1 set to 'eliminated' Status of phase 1 is: ELIMINATED Status of phase 1 set to 'entered' Status of phase 1 is: ENTERED
/* Change status of phase constituent #1 of phase #1 */ tqcspc(1, 1, "dormant", &noerr); if (noerr) abortprog(__LINE__,"tqcspc",noerr); printf("Status of constituent 1 of phase 1 set to 'dormant'\n"); /* Get status of phase constituent #1 of phase #1 */ tqgspc(1, 1, dstr, &noerr); if (noerr) abortprog(__LINE__,"tqgspc",noerr); printf("Status of constituent 1 of phase 1 is: %s \n", dstr); /* Change status of phase constituent #1 of phase #1 */ tqcspc(1, 1, "entered", &noerr); if (noerr) abortprog(__LINE__,"tqcspc",noerr); printf("Status of constituent 1 of phase 1 set to 'entered'\n"); /* Get status of phase constituent #1 of phase #1 */ tqgspc(1, 1, dstr, &noerr); if (noerr) abortprog(__LINE__,"tqgspc",noerr); printf("Status of constituent 1 of phase 1 is: %s \n\n", dstr);
Status of constituent 1 of phase 1 set to 'dormant' Status of constituent 1 of phase 1 is: DORMANT Status of constituent 1 of phase 1 set to 'entered' Status of constituent 1 of phase 1 is: ENTERED
printf("\n\nCalculations using global conditions:\n\n"); /* Set equilibrium conditions */ /* When setting equilibrium conditions with tqsetc, the currently set units for the various quantities (pressure, amount, etc.) apply. Thus, in the following call, the incoming amount ("ia") of phase constituent #4 of phase #1 is set to 1.0 gram. */ tqsetc("ia ", 1, 4, 1.0,&numcon,&noerr); if (noerr) abortprog(__LINE__,"tqsetc",noerr); /* Change system unit */ /* Now we change the amount unit back to "mol". This does not affect any previously set values. The amount of phase constituent #4 of phase #1 we set above is still 1.0 gram, and is not changed to 1.0 mol! */ tqcsu("Amount","mol", &noerr); if (noerr) abortprog(__LINE__,"tqcsu",noerr); printf("Amount unit set to mol\n\n");
Calculations using global conditions: Amount unit set to mol
/* Since we changed the amount unit to "mol", the call to tqsetc below inputs 3.0 mol of constituent #12 of phase #1 */ tqsetc("ia ", 1, 12, 3.0,&numcon,&noerr); if (noerr) abortprog(__LINE__,"tqsetc",noerr); /* Now we input 8 mol of constituent #8 of phase #1 */ tqsetc("ia ", 1, 8, 2.0,&numcon,&noerr); if (noerr) abortprog(__LINE__,"tqsetc",noerr); /* Remove equilibrium condition */ /* This call to tqremc demonstrates how the parameter "numcon" is used, which is returned with every call to tqsetc: The previously defined condition is removed again */ tqremc(numcon, &noerr); if (noerr) abortprog(__LINE__,"tqremc",noerr); /* Set temperature */ /* Here tqsetc is used to enter the temperature. Since we didn't change the default temperature unit, it is still Kelvin ("K") */ tqsetc("t ", 0, 0, 1800.0,&numcon,&noerr); if (noerr) abortprog(__LINE__,"tqsetc",noerr); /* Change limit of target variables */ /* The next call demonstrates how tqclim is used. Normally though calls to tqclim are only necessary under special circumstances */ tqclim("plow", 1e-49, &noerr); if (noerr) abortprog(__LINE__,"tqclim",noerr); /* Display present settings */ /* tqshow outputs the conditions which are active at this point */ printf("\n\nCurrently active conditions:\n"); printf("\n\n**** Begin output table produced by tqshow\n"); /* Depending on the type of ChemApp object code used (e.g. statically linked OBJ code vs. dynamic link library), and possibly also depending on the compiler/platform/operating system used, it might be necessary to use fflush(NULL) to flush the output buffers in order to synchronise the output of the standard C calls and the output through FORTRAN units: */ fflush(NULL); tqshow(&noerr); fflush(NULL); printf("\n**** End output table produced by tqshow\n\n\n"); if (noerr) abortprog(__LINE__,"tqshow",noerr); printf("\n\n");
Currently active conditions: **** Begin output table produced by tqshow TARGET LIMITS: Pressure/bar : 1.00000E-49 1.00000E+07 Volume/dm3 : 1.00000E-07 1.00000E+50 Temperature/K: 298.15 6000.00 T = 1800.00000000 K P = 1 bar STREAM CONSTITUENT CO/GAS/ AMOUNT/mol = 3.57010253334476E-02 Si3/GAS/ AMOUNT/mol = 3.00000000000000E+00 INACTIVE ELECTRON CONSTRAINT **** End output table produced by tqshow
/* Calculate equilibrium */ /* A simple call to tqce or tqcel is sufficient to calculate the equilibrium. All input parameters to tqce and tqcel are only significant if target calculations are performed. */ darray2[0]=0.0; tqce(" ",0,0,darray2,&noerr); if (noerr) abortprog(__LINE__,"tqce",noerr); /* Calculate and list equilibrium */ /* tqcel calculates the equilibrium just like tqce, with the only difference that a ChemSage-type result table is written to the file/unit associated with 'LIST' (see tqgio/tqcio)*/ printf("\n\n**** Begin output table produced by tqcel\n"); fflush(NULL); tqcel(" ",0,0,darray2,&noerr); fflush(NULL); printf("\n**** End output table produced by tqcel\n\n\n"); if (noerr) abortprog(__LINE__,"tqcel",noerr);
**** Begin output table produced by tqcel
T = 1800 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
CO/GAS/ 3.5701E-02
Si3/GAS/ 3.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
SiO 0.0000E+00 9.1172E-01 4.1405E-02
CO 0.0000E+00 8.8214E-02 4.0062E-03
Si 0.0000E+00 6.7137E-05 3.0490E-06
CO2 0.0000E+00 1.1335E-06 5.1478E-08
Si2C 0.0000E+00 7.0003E-07 3.1792E-08
Si2 0.0000E+00 4.5031E-07 2.0451E-08
SiO2 0.0000E+00 1.1236E-07 5.1026E-09
Si3 0.0000E+00 1.0170E-07 4.6188E-09
SiC 0.0000E+00 7.3913E-12 3.3567E-13
O 0.0000E+00 7.2737E-12 3.3033E-13
C 0.0000E+00 1.2567E-13 5.7072E-15
O2 0.0000E+00 1.6031E-16 7.2803E-18
C2 0.0000E+00 9.7381E-17 4.4225E-18
C3 0.0000E+00 4.0495E-17 1.8391E-18
O3 0.0000E+00 8.7034E-33 3.9526E-34
TOTAL: 0.0000E+00 1.0000E+00 4.5415E-02
mol ACTIVITY
Si 8.9464E+00 1.0000E+00
SiC 3.5701E-02 1.0000E+00
SiO2(cristobali) 1.7851E-02 1.0000E+00
SiO2(tridymite) 0.0000E+00 9.9970E-01
SiO2(liquid) 0.0000E+00 9.3907E-01
SiO2(quartz) T 0.0000E+00 9.1491E-01
C 0.0000E+00 2.2526E-02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
2.46520E+02 7.84578E+05 8.41507E+02 -7.30135E+05 0.00000E+00
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
**** End output table produced by tqcel
/* Once an equilibrium has been calculated using tqce/tqcel, subsequent calculations can be performed using tqcen/tqcenl, which speeds up the equilibrium calculation by taking results from the previous calculation as initial estimates */ tqsetc("t ", 0, 0, 1850.0,&numcon,&noerr); if (noerr) abortprog(__LINE__,"tqsetc",noerr); tqcen(" ",0,0,darray2,&noerr); if (noerr) abortprog(__LINE__,"tqcen",noerr); tqsetc("t ", 0, 0, 1900.0,&numcon,&noerr); if (noerr) abortprog(__LINE__,"tqsetc",noerr); /* Similar to tqcel, tqcenl also provides a ChemSage output table */ printf("\n\n**** Begin output table produced by tqcenl\n"); fflush(NULL); tqcenl(" ",0,0,darray2,&noerr); fflush(NULL); printf("\n**** End output table produced by tqcenl\n\n\n"); if (noerr) abortprog(__LINE__,"tqcenl",noerr); /* Change value of input/output option */ /* Redirect the output from tqcel to a file using tqcio with the option "LIST"*/ /* The following call to tqcio will redirect the output of ChemApp subroutines like tqcel and tqshow to the FORTRAN unit number 21. Note that only certain values for the unit number are permitted (see the ChemApp manual entry for tqcio) */ tqcio("LIST", 21, &noerr); if (noerr) abortprog(__LINE__,"tqcio",noerr); /* Note that at this point, no file has been associated with unit number 21 yet! This is done with the subsequent call to tqopen:*/ tqopen("result",21,&noerr); if (noerr) abortprog(__LINE__,"tqopen",noerr); printf("Output of next call to tqcel will be written to " "file \"result\"\n"); /* tqwstr can be used to write user-defined text to the unit numbers associated with "LIST" and "ERROR". This subroutine is especially useful for non-FORTRAN programs. */ tqwstr("LIST", "Output from tqcel (ChemSage result table):", &noerr); if (noerr) abortprog(__LINE__,"tqwstr",noerr); /* Now the ChemSage-type result table output by the following routine is written to the file "result": */ tqcel(" ",0,0,darray2,&noerr); if (noerr) abortprog(__LINE__,"tqcel",noerr); /* Close the file associated with unit number 21 again... */ tqclos(21,&noerr); if (noerr) abortprog(__LINE__,"tqclos",noerr); /* ...and redirect the output of routines like tqcel and tqshow back to unit number 6, which is the default and associated with stdout: */ tqcio("LIST", 6, &noerr); if (noerr) abortprog(__LINE__,"tqcio",noerr); printf("Output of subsequent calls to tqcel and tqshow will go " "to stdout again\n\n");
**** Begin output table produced by tqcenl
T = 1900 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
CO/GAS/ 3.5701E-02
Si3/GAS/ 3.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
SiO 0.0000E+00 8.5125E-01 1.2301E-01
CO 0.0000E+00 1.4866E-01 2.1481E-02
Si 0.0000E+00 8.4025E-05 1.2142E-05
CO2 0.0000E+00 3.7718E-06 5.4503E-07
Si2C 0.0000E+00 1.4416E-06 2.0832E-07
Si2 0.0000E+00 7.4859E-07 1.0817E-07
SiO2 0.0000E+00 2.7664E-07 3.9975E-08
Si3 0.0000E+00 1.6036E-07 2.3173E-08
SiC 0.0000E+00 3.6804E-11 5.3182E-12
O 0.0000E+00 2.9557E-11 4.2711E-12
C 0.0000E+00 7.5814E-13 1.0955E-13
O2 0.0000E+00 1.3988E-15 2.0213E-16
C2 0.0000E+00 1.3301E-15 1.9220E-16
C3 0.0000E+00 7.2884E-16 1.0532E-16
O3 0.0000E+00 6.6967E-31 9.6770E-32
TOTAL: 0.0000E+00 1.0000E+00 1.4450E-01
mol ACTIVITY
Si 8.9464E+00 1.0000E+00
SiC 3.5701E-02 1.0000E+00
SiO2(cristobali) 1.7851E-02 1.0000E+00
SiO2(tridymite) 0.0000E+00 9.9918E-01
SiO2(liquid) 0.0000E+00 9.7125E-01
SiO2(quartz) T 0.0000E+00 9.0635E-01
C 0.0000E+00 3.4700E-02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
2.46541E+02 8.09231E+05 8.54837E+02 -8.14958E+05 0.00000E+00
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
**** End output table produced by tqcenl
Output of next call to tqcel will be written to file "result"
Output of subsequent calls to tqcel and tqshow will go to stdout again
/* Get and display results */ /* First, we get the fraction of the first phase constituent of the first phase. We get also its name with tqgnpc, this time we use the string variable "dstr2" which we declared using the macro "TQSTRING". Since the current amount unit is "mol", we get the "mole fraction" (as opposed to "mass fraction") */ tqgnsc(1, dstr2, &noerr); if (noerr) abortprog(__LINE__,"tqgnsc",noerr); tqgetr("xp ", 1, 1, &d1, &noerr); if (noerr) abortprog(__LINE__,"tqgetr",noerr); printf("Mole fraction of system component %s in the GAS phase: %f\n\n", dstr2, d1);
Mole fraction of system component C in the GAS phase: 0.074334
/* Now get the equilibrium amount of the same constituent */ tqgetr("a ",1,1,&d1,&noerr); if (noerr) abortprog(__LINE__,"tqgetr",noerr); printf("Equilibrium amount of %s in the GAS phase: %f\n\n", dstr2, d1);
Equilibrium amount of C in the GAS phase: 0.000000
/* Note that the result is zero, because the gas phase is not considered stable. The reason is that we are using the default ambient pressure of 1 bar for the calculations, and from the result table produced with the previous call to tqcel or the subsequent call to tqgetr we see that the activity of the gas phase is less than unity, which means it is not considered stable, and thus the equilibrium amounts of all its phase constituents are zero. */ /* Using the option "ac", tqgetr retrieves activities, in this case the activity (fugacity) of the gas phase */ tqgetr("ac ",1, 0, &d1, &noerr); if (noerr) abortprog(__LINE__,"tqgetr",noerr); printf("Activity of the GAS phase: %f\n\n", d1);
Activity of the GAS phase: 0.144503
/* Now tqgetr is used in a way that causes it to return an array. In this case, we are going to retrieve an array that contains the fugacities of all constituents of the gas phase. First, we need an array that has enough room for all the results.*/ /* With an earlier call to tqnopc we determined the number of constituents in the gas phase and stored it in npcgas. Thus we can use this number to allocate enough memory for the array:*/ if (!(darray = calloc(npcgas, sizeof(DB)))) { printf("Not enough memory available for dynamic allocation!\n"); exit(1); } /* Get the fugacities:*/ tqgetr("ac", 1, -1, darray, &noerr); if (noerr) abortprog(__LINE__,"tqgetr",noerr); /* Print them out, together with the names of the constituents (note that these values correspond to the last column of the result table previously output with tqcel):*/ printf("\nFugacities of all gas phase constituents:\n"); /* Note that the 0th element of the array contains the fugacity of the 1st constituent of the gas phase. That's why the call to tqgnpc has to use "lint+1" instead of "lint"! */ for (lint = 0; lint < npcgas; lint++) { tqgnpc(1, lint+1, dstr, &noerr); if (noerr) abortprog(__LINE__,"tqgnpc",noerr); printf("%5s: %g\n", dstr, darray[lint]); }
Fugacities of all gas phase constituents:
C: 1.09553e-13
C2: 1.92202e-16
C3: 1.0532e-16
CO: 0.0214813
CO2: 5.45033e-07
O: 4.27106e-12
O2: 2.02127e-16
O3: 9.67695e-32
Si: 1.21418e-05
Si2: 1.08174e-07
Si2C: 2.08321e-07
Si3: 2.31727e-08
SiC: 5.31822e-12
SiO: 0.123008
SiO2: 3.99754e-08
printf("\n\n"); /* Recycle the memory */ free(darray); /* Get thermodynamic data of a phase constituent */ tqgdpc("G", 1, 1, &d1, &noerr); if (noerr) abortprog(__LINE__,"tqgdpc",noerr); printf("The (dimensionless) value of G for " "constituent 1 of phase 1 is: %f\n\n\n", d1);
The (dimensionless) value of G for constituent 1 of phase 1 is: 23.825850
/* Note that when retrieving CP, H, S, or G using tqgdpc, the values are returned "dimensionless", which means they might have to be multiplied by R*T. Since the default amount unit is mol, results are returned for 1 mol. Note also that care has to be taken if a temperature unit different from Kelvin has been used. Refer to the manual entry of tqgdpc for a more detailed example. */ /* Check if we are working with the 'light' version. If we do, omit the following phase target calculation(s). */ tqlite(&islite, &noerr); if (islite) { printf("*** Phase target calculations have been omitted here,\n" "*** since they are not possible with the\n" "*** 'light' version of ChemApp.\n\n"); } else { /* Perform a target calculation. This example demonstrates how to have ChemApp find the temperature at which a certain phase becomes stable. */ /* We want to have ChemApp calculate the temperature at which the phase "SiO2(liquid)" becomes stable. First, determine the index number of this phase (note that it is allowed to abbreviate its name, as long as it is unambiguous):*/ tqinp("SiO2(liq", &lint, &noerr); if (noerr) abortprog(__LINE__,"tqinp",noerr); /* Next, define the formation target :*/ tqsetc("a", lint, 0, 0.0, &numcon, &noerr); if (noerr) abortprog(__LINE__,"tqsetc",noerr); /* Then call tqcel to calculate the equilibrium, and pass it the information about the target variable (we want ChemApp to vary the _temperature_ until the phase is stable): */ printf("Performing a phase target calculation\n" "Phase target: formation of SiO2(liquid)\n" "Target variable: Temperature\n"); darray2[0] = 2000.0; printf("\n\n**** Begin output table produced by tqcel\n"); fflush(NULL); tqcel("t",0,0,darray2,&noerr); fflush(NULL); printf("\n**** End output table produced by tqcel\n\n\n");
Performing a phase target calculation
Phase target: formation of SiO2(liquid)
Target variable: Temperature
**** Begin output table produced by tqcel
*T = 1995.99 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
CO/GAS/ 3.5701E-02
Si3/GAS/ 3.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
SiO 0.0000E+00 7.7442E-01 3.1442E-01
CO 0.0000E+00 2.2547E-01 9.1542E-02
Si 0.0000E+00 9.8748E-05 4.0093E-05
CO2 0.0000E+00 1.0313E-05 4.1873E-06
Si2C 0.0000E+00 2.5977E-06 1.0547E-06
Si2 0.0000E+00 1.1230E-06 4.5595E-07
SiO2 0.0000E+00 5.8253E-07 2.3651E-07
Si3 0.0000E+00 2.2928E-07 9.3089E-08
SiC 0.0000E+00 1.4260E-10 5.7898E-11
O 0.0000E+00 9.6352E-11 3.9120E-11
C 0.0000E+00 3.4767E-12 1.4116E-12
C2 0.0000E+00 1.2342E-14 5.0109E-15
O2 0.0000E+00 8.8186E-15 3.5804E-15
C3 0.0000E+00 8.5734E-15 3.4809E-15
O3 0.0000E+00 2.7760E-29 1.1271E-29
TOTAL: 0.0000E+00 1.0000E+00 4.0601E-01
mol ACTIVITY
Si 8.9464E+00 1.0000E+00
SiC 3.5701E-02 1.0000E+00
SiO2(cristobali) 1.7851E-02 1.0000E+00
SiO2(liquid) 0.0000E+00 1.0000E+00
SiO2(tridymite) T 0.0000E+00 9.9861E-01
SiO2(quartz) T 0.0000E+00 8.9882E-01
C 0.0000E+00 5.0396E-02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
2.46559E+02 8.32897E+05 8.66988E+02 -8.97600E+05 0.00000E+00
Data on 2 constituents marked with 'T' are extrapolated outside their valid
temperature range
**** End output table produced by tqcel
if (noerr) abortprog(__LINE__,"tqcel",noerr); darray2[0] = 0.0; /* Note that in the resulting ChemSage table, the temperature is marked with an asterisk, meaning that it has been calculated by ChemApp, and the activity of SiO2(liquid) is unity, whereas its equilibrium amount is zero, meaning it just forms at this temperature*/ /* Retrieve the calculated temperature:*/ tqgetr("t", 0, 0, &d1, &noerr); if (noerr) abortprog(__LINE__,"tqgetr",noerr); printf("Calculated formation temperature of SiO2(liquid): %g\n\n", d1);
Calculated formation temperature of SiO2(liquid): 1995.99
/* One-dimensional phase mapping calculations. The example below demonstrates how to locate all phase transitions in a given temperature range. */ /* One-dimensional phase mapping calculations are only possible with version 3.x and later of ChemApp. */ printf("Performing one-dimensional phase mapping calculations\n"); /* Initialise the variable to zero that holds the number of times tqmap found results */ resno = 0; /* Remove all previous conditions */ tqremc(-2, &noerr); if (noerr) abortprog(__LINE__,"tqremc",noerr); /* Determine the index number for the phase SiO2(quartz) */ tqinp("SiO2(quartz)", &lint, &noerr); if (noerr) abortprog(__LINE__,"tqinp",noerr); /* Enter one mol of SiO2 */ tqsetc("IA", lint, 0, 1.0, &numcon, &noerr); if (noerr) abortprog(__LINE__,"tqsetc",noerr); /* The temperature search interval is supposed to range from 300 to 3000 K: */ darray2[0] = 300.0; darray2[1] = 3000.0; /* First call to tqmap, note the "f" ("first") in the option parameter */ tqmap("tf", 0, 0, darray2, &icont, &noerr); if (noerr) abortprog(__LINE__,"tqmap",noerr); /* The variable resno keeps track of the number of times we call tqmap: */ resno++; /* Retrieve and print the temperature, which we know is darray2[0]: */ tqgetr("t", 0, 0, &d1, &noerr); if (noerr) abortprog(__LINE__,"tqgetr",noerr); printf("*** Lower interval boundary: %g K\n", d1); /* For as long as icont is positive, we need to make further calls to tqmap */ while (icont) { /* tqmap is called again. Note the "n" ("next") in the option parameter. If we are at the first phase boundary (resno is still 2), we call tqmapl for a change to produce a ChemSage output table... */ if (resno == 2) { printf("*** ChemSage result table " "for the first phase boundary found:\n"); fflush(NULL); tqmapl("tn", 0, 0, darray2, &icont, &noerr); fflush(NULL); if (noerr) abortprog(__LINE__,"tqmapl",noerr); printf("\n"); /* ...otherwise we just call tqmap: */ } else { tqmap("tn", 0, 0, darray2, &icont, &noerr); if (noerr) abortprog(__LINE__,"tqmap",noerr); } resno++; /* Get the temperature... */ tqgetr("t", 0, 0, &d1, &noerr); if (noerr) abortprog(__LINE__,"tqgetr",noerr); /* ...and print the entry for the result table. If we have called tqmap twice already, we know that we found a phase boundary. If not, we have retrieved the temperature value of the upper interval boundary (darray2[1]): */ if (resno > 2) { printf("*** Phase boundary found at %g K\n", d1); } else { printf("*** Upper interval boundary: %g K\n", d1); } }
Performing one-dimensional phase mapping calculations
*** Lower interval boundary: 300 K
*** Upper interval boundary: 3000 K
*** ChemSage result table for the first phase boundary found:
*T = 1140.10 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol
SiO2(quartz) 1.0000E+00
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
SiO 0.0000E+00 6.2502E-01 4.4829E-16
O2 0.0000E+00 2.5088E-01 1.7994E-16
O 0.0000E+00 1.2327E-01 8.8415E-17
SiO2 0.0000E+00 8.3208E-04 5.9681E-19
Si 0.0000E+00 4.4651E-15 3.2026E-30
O3 0.0000E+00 2.4636E-19 1.7670E-34
Si2 0.0000E+00 5.5659E-36 3.9921E-51
Si3 0.0000E+00 9.0161E-54 6.4667E-69
TOTAL: 0.0000E+00 1.0000E+00 7.1725E-16
mol ACTIVITY
SiO2(quartz) 1.0000E+00 1.0000E+00
SiO2(tridymite) 0.0000E+00 1.0000E+00
SiO2(cristobali) 0.0000E+00 9.9398E-01
SiO2(liquid) 0.0000E+00 6.4439E-01
Si 0.0000E+00 2.5802E-17
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
7.04990E+01 -8.55398E+05 1.25413E+02 -9.98382E+05 0.00000E+00
*** Phase boundary found at 1140.1 K
*** Phase boundary found at 1738.28 K
*** Phase boundary found at 1995.99 K
/* With the above example the temperatures of all phase boundaries in a system which contains 1 mol of SiO2 have been calculated. Thus the phase boundaries determined reflect the stability ranges of the various modifications of SiO2. Also note that the first two temperatures determined are _no_ phase boundaries, but the lower and upper limit of the search interval (darray2[0] and darray2[1]). */ } /* Since from now on we will be using streams instead of global conditions, we first remove all conditions and targets set up to this point. Note that using a "-1" instead of the "-2" in the call to tqremc would also reset ChemApp to default units and values.*/ tqremc(-2, &noerr); if (noerr) abortprog(__LINE__,"tqremc",noerr); printf("\n\nCalculations using streams:\n\n"); /* Set name and temperature for a stream via the array tp, which will be passed to tqsttp when each of the streams is created. */ tp[0] = 1000.0; tp[1] = 1.0; /* Create 3 streams: */ tqsttp("stream1", tp, &noerr); if (noerr) abortprog(__LINE__,"tqsttp",noerr); tqsttp("stream2", tp, &noerr); if (noerr) abortprog(__LINE__,"tqsttp",noerr); tqsttp("stream3", tp, &noerr); if (noerr) abortprog(__LINE__,"tqsttp",noerr); /* Set constituent amounts for each stream */ tqstca("stream1", 1, 4, 1.0,&noerr); if (noerr) abortprog(__LINE__,"tqstca",noerr); tqstca("stream2", 1, 12, 3.0,&noerr); if (noerr) abortprog(__LINE__,"tqstca",noerr); tqstca("stream3", 1, 8, 2.0,&noerr); if (noerr) abortprog(__LINE__,"tqstca",noerr); /* Remove the last stream */ tqstrm("stream3", &noerr); if (noerr) abortprog(__LINE__,"tqstrm",noerr); /* Set equilibrium temperature */ tqstec("t ", 0, 1800.0, &noerr); if (noerr) abortprog(__LINE__,"tqstec",noerr); /* Calculate and list equilibrium */ printf("\n\n**** Begin output table produced by tqcel\n"); fflush(NULL); tqcel(" ",0,0,darray2,&noerr); fflush(NULL); printf("\n**** End output table produced by tqcel\n\n\n");
Calculations using streams:
**** Begin output table produced by tqcel
T = 1800 K
P = 1 bar
V = 0 dm3
STREAM CONSTITUENTS AMOUNT/mol TEMPERATURE/K PRESSURE/bar STREAM
CO/GAS/ 1.0000E+00 1000.00 1.0000E+00 2
Si3/GAS/ 3.0000E+00 1000.00 1.0000E+00 3
EQUIL AMOUNT MOLE FRACTION FUGACITY
PHASE: GAS mol bar
SiO 0.0000E+00 9.1172E-01 4.1405E-02
CO 0.0000E+00 8.8214E-02 4.0062E-03
Si 0.0000E+00 6.7137E-05 3.0490E-06
CO2 0.0000E+00 1.1335E-06 5.1478E-08
Si2C 0.0000E+00 7.0003E-07 3.1792E-08
Si2 0.0000E+00 4.5031E-07 2.0451E-08
SiO2 0.0000E+00 1.1236E-07 5.1026E-09
Si3 0.0000E+00 1.0170E-07 4.6188E-09
SiC 0.0000E+00 7.3913E-12 3.3567E-13
O 0.0000E+00 7.2737E-12 3.3033E-13
C 0.0000E+00 1.2567E-13 5.7072E-15
O2 0.0000E+00 1.6031E-16 7.2803E-18
C2 0.0000E+00 9.7381E-17 4.4225E-18
C3 0.0000E+00 4.0495E-17 1.8391E-18
O3 0.0000E+00 8.7034E-33 3.9526E-34
TOTAL: 0.0000E+00 1.0000E+00 4.5415E-02
mol ACTIVITY
Si 7.5000E+00 1.0000E+00
SiC 1.0000E+00 1.0000E+00
SiO2(cristobali) 5.0000E-01 1.0000E+00
SiO2(tridymite) 0.0000E+00 9.9970E-01
SiO2(liquid) 0.0000E+00 9.3907E-01
SiO2(quartz) T 0.0000E+00 9.1491E-01
C 0.0000E+00 2.2526E-02
********************************************************************
DELTA Cp DELTA H DELTA S DELTA G DELTA V
J.K-1 J J.K-1 J dm3
********************************************************************
7.76667E+01 -1.68039E+06 -3.77532E+02 -2.00297E+06 -3.32581E+02
********************************************************************
Cp H S G V
J.K-1 J J.K-1 J dm3
********************************************************************
2.93963E+02 2.64115E+05 8.75143E+02 -1.31114E+06 0.00000E+00
Data on 1 constituent marked with 'T' are extrapolated outside their valid
temperature range
**** End output table produced by tqcel
if (noerr) abortprog(__LINE__,"tqcel",noerr); /* Get thermodynamic properties of a stream (note that as of version 3.2.0 of ChemApp, tqstxp can also be called _before_ the equilibrium calculation) */ tqstxp("stream1", "H", &d1, &noerr); if (noerr) abortprog(__LINE__,"tqstxp",noerr); printf("Enthalpy of \"stream1\": %g\n\n", d1); printf("\nDemonstrating the use of ChemApp " "subroutines involving sublattices.\n\n"); /* To demonstrate the subroutines dealing with sublattices, a different data-file (subl-ex.dat) needs to be loaded. This data-file contains an extract of the system Co-Cr-Fe: the phase SIGMA:30, which is modelled according to the sublattice formalism, and the BCC phase, described by a Redlich-Kister polynomial. Both phases are each included twice, to account for miscibility gaps. */ tqopna("subl-ex.dat", unitno, &noerr); if (noerr) abortprog(__LINE__,"tqopna",noerr); /* Read data-file */ tqrfil(&noerr); if (noerr) abortprog(__LINE__,"tqrfil",noerr); /* Close data-file */ tqclos(unitno, &noerr); if (noerr) abortprog(__LINE__,"tqclos",noerr); /* The first of the two identical copies of the SIGMA:30 phase, which ChemApp calls SIGMA:30#1, will be investigated with respect to the number of sublattices, the number of sublattice constituents on each sublattice, and the names of the sublattice constituents. */ /* Get the index number for the phase SIGMA:30#1 */ strcpy(pname, "SIGMA:30#1"); tqinp(pname, &indexp, &noerr); if (noerr) abortprog(__LINE__,"tqinp",noerr); /* Get the number of sublattices */ tqnosl(indexp, &nsl, &noerr); if (noerr) abortprog(__LINE__,"tqnosl",noerr); printf("%s has %li sublattices\n",pname, nsl);
Enthalpy of "stream1": -88842.4 Demonstrating the use of ChemApp subroutines involving sublattices. SIGMA:30#1 has 3 sublattices
/* Loop over all sublattices */ for (indexl = 1; indexl<=nsl; indexl++) { /* Get the number of sublattice constituents */ tqnolc(indexp, indexl, &nslcon, &noerr); if (noerr) abortprog(__LINE__,"tqnolc",noerr); printf("Sublattice %li has %li constituents with the following names:\n", indexl, nslcon); /* Get the name of each sublattice constituent */ for (indexc = 1; indexc<=nslcon; indexc++) { tqgnlc(indexp, indexl ,indexc, cname, &noerr); if (noerr) abortprog(__LINE__,"tqgnlc",noerr); /* The reverse (getting the index number for the name of the sublattice constituent just retrieved), is rather superfluous here, and only used to demonstrate the call to tqinlc: */ tqinlc(cname, indexp, indexl, &lint, &noerr); if (noerr) abortprog(__LINE__,"tqinlc",noerr); printf(" %li: %s\n", lint, cname); } }
Sublattice 1 has 2 constituents with the following names: 1: Co 2: Fe Sublattice 2 has 1 constituents with the following names: 1: Cr Sublattice 3 has 3 constituents with the following names: 1: Co 2: Cr 3: Fe
/* Set the temperature to 1000 K */ tqsetc("T", 0, 0, 1000.0, &numcon, &noerr); if (noerr) abortprog(__LINE__,"tqsetc",noerr); /* Set the incoming amounts to 0.25 mol Co, 0.25 mol Cr, and 0.5 mol Fe */ tqinsc("Co", &indexc, &noerr); if (noerr) abortprog(__LINE__,"tqinsc",noerr); tqsetc("ia", 0, indexc, 0.25, &numcon, &noerr); if (noerr) abortprog(__LINE__,"tqsetc",noerr); tqinsc("Cr", &indexc, &noerr); if (noerr) abortprog(__LINE__,"tqinsc",noerr); tqsetc("ia", 0, indexc, 0.25, &numcon, &noerr); if (noerr) abortprog(__LINE__,"tqsetc",noerr); tqinsc("Fe", &indexc, &noerr); if (noerr) abortprog(__LINE__,"tqinsc",noerr); tqsetc("ia", 0, indexc, 0.50, &numcon, &noerr); if (noerr) abortprog(__LINE__,"tqsetc",noerr); printf("\nCalculate the equilibrium, " "get information on the stable sublattice phases.\n\n"); /* Calculate the equilibrium */ darray2[0]=0.0; tqce(" ", 0, 0, darray2, &noerr); if (noerr) abortprog(__LINE__,"tqce",noerr); /* Get information on the stable sublattice phases. */ /* Loop over all phases, and if a phase is stable and a sublattice phase, print information about the sublattices */ tqnop(&nphase, &noerr); if (noerr) abortprog(__LINE__,"tqnop",noerr); for (indexp = 1; indexp<=nphase; indexp++) { /* Check if the phase is stable, otherwise we don't need to consider it */ tqgetr("a", indexp, 0, &d1, &noerr); if (noerr) abortprog(__LINE__,"tqgetr",noerr); if (d1 > 0) { /* Get the name and model of the phase */ tqgnp(indexp, pname, &noerr); if (noerr) abortprog(__LINE__,"tqgnp",noerr); tqmodl(indexp, mname, &noerr); if (noerr) abortprog(__LINE__,"tqmodl",noerr); /* If the model name of the phase starts with 'SUB' we have a sublattice phase */ if (strncmp(mname, "SUB", 3) == 0) { /* Print a header similar to the one in the ChemSage output table */ printf("Mole fraction of the sublattice constituents in %s\n", pname); /* Get the number of sublattices */ tqnosl(indexp, &nsl, &noerr); if (noerr) abortprog(__LINE__,"tqnosl",noerr); /* Loop over all sublattices */ for (indexl=1; indexl<=nsl; indexl++) { printf("Sublattice %li\n", indexl); /* Get the number of sublattice constituents */ tqnolc(indexp, indexl, &nslcon, &noerr); if (noerr) abortprog(__LINE__,"tqnolc",noerr); for (indexc=1; indexc<=nslcon; indexc++) { /* Get the name of each sublattice constituent... */ tqgnlc(indexp, indexl, indexc, cname, &noerr); if (noerr) abortprog(__LINE__,"tqgnlc",noerr); /* ... and its mole fraction in the sublattice */ tqgtlc(indexp, indexl, indexc, &d1, &noerr); if (noerr) abortprog(__LINE__,"tqgtlc",noerr); printf("%-25s %f\n", cname, d1); } } } } } printf("\n");
Calculate the equilibrium, get information on the stable sublattice phases. Mole fraction of the sublattice constituents in SIGMA:30#1 Sublattice 1 Co 0.542377 Fe 0.457623 Sublattice 2 Cr 1.000000 Sublattice 3 Co 0.109259 Cr 0.691920 Fe 0.198821
/* The following section demonstrates the use of subroutines that were introduced in ChemApp V4.0.0 to support "transparent data-files" */ /* Retrieve the licensee's user ID */ tqgtid(id, &noerr); if (noerr) abortprog(__LINE__,"tqgtid",noerr); /* Retrieve the licensee's name */ tqgtnm(name, &noerr); if (noerr) abortprog(__LINE__,"tqgtnm",noerr); /* Retrieve the program ID */ tqgtpi(pid, &noerr); if (noerr) abortprog(__LINE__,"tqgtpi",noerr); /* Print all three */ printf("Licensee's user ID: %s\n", id); printf("Licensee's name : %s\n", name); printf("Program ID : %s\n\n\n", pid);
Licensee's user ID: 5001 Licensee's name : GTT - Technologies Program ID : CAFU
/* The following pieces of information are only meaningful if a version of ChemApp is used that requires a dongle (hardware key). Get the HASP dongle type and id */ tqgthi(haspt, &haspid, &noerr); if (noerr) abortprog(__LINE__,"tqgthi",noerr); /* Get the ChemApp license expiration date (month and year) */ tqgted(&edmon, &edyear, &noerr); if (noerr) abortprog(__LINE__,"tqgted",noerr); printf("ChemApp license expiration date (month/year): %li/%li\n\n\n", edmon, edyear);
ChemApp license expiration date (month/year): 12/2025
/* Print info if HASP dongle is used: */ if (haspid) { printf("HASP dongle type : %s\n", haspt); printf("HASP dongle id : %li\n\n\n", haspid); } else { printf("This ChemApp version does not require a " "HASP hardware key (dongle)\n\n\n"); }
This ChemApp version does not require a HASP hardware key (dongle)
/* Get the default unit number associated with error output */ tqgio("ERROR", &errorunit, &noerr); if (noerr) abortprog(__LINE__,"tqgio",noerr); /* Turn off automatic error reporting for the next call to tqcio. */ tqcio("ERROR", 0, &noerr); if (noerr) abortprog(__LINE__,"tqcio",noerr); /* The following section is only executed if the file "cosiex.cst", a sample thermochemical data-file in transparent format, is present. It is not included in regular distributions of ChemApp */ tqopnt("cosiex.cst",unitno,&lint); /* Turn automatic error reporting back on. */ tqcio("ERROR", errorunit, &noerr); if (noerr) abortprog(__LINE__,"tqcio",noerr); if (lint) { printf("Skipping transparent file subroutines, since file " "'cosiex.cst' cannot be read\n"); } else { /* Read data-file */ tqrcst(&noerr); if (noerr) abortprog(__LINE__,"tqrcst",noerr); /* Close data-file */ /* Again, the routine for closing the data-file is not a standard C library routine like fclose, but a special one to make sure the file that was previously opened under the unit number specified is closed. Both tqopnb and tqclos are not actually a part of ChemApp itself, but of ChemApp's C interface. */ tqclos(unitno,&noerr); if (noerr) abortprog(__LINE__,"tqclos",noerr); /* Once the transparent data-file has been read, information on its header can be retrieved.*/ tqgtrh(&tfhver, tfhnwp, tfhvnw, tfhnrp, tfhvnr, tfhdtc, tfhdte, tfhid, tfhusr, tfhrem, &noerr); if (noerr) abortprog(__LINE__,"tqgtrh",noerr); printf("Version number of the transparent file header format: %li\n", tfhver); printf("Name of the program which wrote the data-file: %s\n", tfhnwp); printf("Version number of the writing program: %li.%li.%li\n", tfhvnw[0],tfhvnw[1],tfhvnw[2]); printf("Programs which are permitted to read the data-file: %s\n", tfhnrp); printf("Min. version number of the reading program: %li.%li.%li\n", tfhvnr[0],tfhvnr[1],tfhvnr[2]); printf("File was created on %li/%02li/%02li %02li:%02li:%02li \n", tfhdtc[0],tfhdtc[1],tfhdtc[2],tfhdtc[3],tfhdtc[4],tfhdtc[5]); printf("File will expire on %li/%02li/%02li %02li:%02li:%02li \n", tfhdte[0],tfhdte[1],tfhdte[2],tfhdte[3],tfhdte[4],tfhdte[5]); printf("Licensee's user ID(s): %s\n", tfhid); printf("Licensee's name: %s\n", tfhusr); printf("Remarks : %s\n", tfhrem); }
Version number of the transparent file header format: 1 Name of the program which wrote the data-file: FactSage Version number of the writing program: 5.0.0 Programs which are permitted to read the data-file: CAFU,CALI Min. version number of the reading program: 4294967295.4294967295.4294967295 File was created on 2015/09/09 15:13:140724603453492 File will expire on 2036/12/31 12:00:140724603453440 Licensee's user ID(s): ???? Licensee's name: GTT - Technologies Remarks : For use with ChemApp example programs
printf("\n\nEnd of output from cademo1.\n"); return 0; }
End of output from cademo1.
14 Example data-files and their contents
The following sections contain the example thermochemical data-files which are part of the ChemApp distribution (see also Example thermochemical data-files).
For a better overview on what is inside each data-file, a directory of contents is presented first, which includes a list of system components, phases, and phase constituents of mixture phases. Below each directory, the data-file itself is displayed. For a description of the structure of a ChemApp data-file, see Thermochemical Data-files - Structure and Handling.
The following data-files are included in this chapter:
14.1 COSI.DAT
Directory
Contents of data-file cosi.dat System components: 1: C 2: O 3: Si Phases: 1: GAS 2: C 3: Si 4: SiC 5: SiO2(quartz) 6: SiO2(tridymite) 7: SiO2(cristobalite) 8: SiO2(liquid) Phase constituents of mixture phases: Phase: GAS Model: IDMX Status: ENTERED 1: C 2: C2 3: C3 4: CO 5: CO2 6: O 7: O2 8: O3 9: Si 10: Si2 11: Si2C 12: Si3 13: SiC 14: SiO 15: SiO2
Data-file
C-O-Si See commentary below ! GTT Nov.1996
3 1 15 7
C O Si
12.0110 15.9994 28.0860
6 1 2 3 4 5 6
1 1
GAS
IDMX
C
1 2 1.0 0.0 0.0
2000.0000 710457.00 -18.971944 -20.769400 -.27196000E-04
0.00000000 0.00000000
6000.0000 711696.00 -28.661644 -19.489100 -.35773200E-03
0.00000000 0.00000000
C2
1 1 2.0 0.0 0.0
6000.0000 826673.00 1.8085362 -30.685500 -.23765100E-02
0.71128000E-07 -508774.00
C3
1 1 3.0 0.0 0.0
4000.0000 774485.00 130.77556 -49.887100 -.35584900E-02
0.17642500E-06 602078.00
CO
1 1 1.0 1.0 0.0
3000.0000 -119348.00 -5.9053738 -28.409400 -.20501600E-02
0.00000000 23012.000
CO2
1 1 1.0 2.0 0.0
3000.0000 -409930.00 89.365756 -44.141200 -.45187200E-02
0.00000000 426768.00
O
1 2 0.0 1.0 0.0
2000.0000 243275.00 -21.813244 -20.874000 0.25104000E-04
0.00000000 -48743.600
3000.0000 243168.00 -21.880544 -20.853100 0.00000000
0.00000000 0.00000000
O2
1 2 0.0 2.0 0.0
3000.0000 -9679.1000 -2.3147038 -29.957400 -.20920000E-02
0.00000000 83680.000
6000.0000 -15637.800 29.763356 -34.045200 -.14204700E-02
0.36958700E-07 2686130.0
O3
1 1 0.0 3.0 0.0
2273.0000 125910.00 67.380256 -44.346200 -.77968800E-02
0.72452900E-06 430534.00
Si
1 1 0.0 0.0 1.0
3000.0000 445338.00 -36.090244 -19.815400 -.50208000E-03
0.00000000 -100416.00
Si2
1 1 0.0 0.0 2.0
3000.0000 575592.00 43.897456 -40.120400 -.22572700E-02
0.22872500E-06 328235.00
Si2C
1 1 1.0 0.0 2.0
6000.0000 517805.00 112.12056 -52.170300 -.31317200E-02
0.52300000E-07 285767.00
Si3
1 1 0.0 0.0 3.0
3000.0000 616323.00 138.37556 -60.178500 -.79286800E-03
0.00000000 243300.00
SiC
1 1 1.0 0.0 1.0
6000.0000 715014.00 25.751956 -37.229200 -.73220000E-03
0.21617300E-07 -973408.00
SiO
1 2 0.0 1.0 1.0
2273.0000 -110346.00 -8.3075638 -29.823600 -.41191500E-02
0.38004700E-06 103136.00
6000.0000 -130585.00 101.41956 -44.358800 0.14267400E-02
-.62760000E-07 6301100.0
SiO2
1 1 0.0 2.0 1.0
6000.0000 -325105.00 154.83356 -56.157600 -.15187900E-02
0.58576000E-07 604170.00
C
1 3 1.0 0.0 0.0
800.00000 -1153.5000 -12.583000 3.0258000 -.23742000E-01
0.39214000E-05 22809.000
1900.0000 -12380.000 139.22000 -20.022000 -.24390000E-02
0.19766000E-06 1066600.0
6000.0000 -17431.000 178.95000 -25.499000 0.98209000E-04
-.15854000E-07 1897800.0
Si
7 2 0.0 0.0 1.0
0.00000000 18.819600
1685.0000 22.823720 0.38576480E-02 0.00000000 -353966.40
50208.000
3492.0000 27.196000 0.00000000 0.00000000 0.00000000
SiC
1 1 1.0 0.0 1.0
3259.0000 -91845.000 280.43000 -42.593100 -.41798200E-02
0.21198900E-06 829687.00
SiO2(quartz)
4 3 0.0 2.0 1.0
373.00000 -935388.53 536.02533 -80.011990 0.00000000
0.00000000 1773342.0
3 0.00000000 4.0 -961.10400 0.5 -81928064. -2.0
848.00000 -935486.57 537.07568 -80.011990 -.42200108E-02
0.75354500E-05 1773342.0
3 -.50458704E-08 4.0 -961.10400 0.5 -81928064. -2.0
1744.2500 -933315.35 533.27853 -80.011990 0.00000000
0.00000000 1773342.0
3 0.00000000 4.0 -961.10400 0.5 -81928064. -2.0
SiO2(tridymite)
4 2 0.0 2.0 1.0
390.15000 -946102.84 500.41936 -75.372670 -.91038535E-01
0.20563620E-03 2979047.5
3 -.17418275E-06 4.0 0.00000000 0.5 -.15970769E+09 -2.0
1991.2800 -944111.17 480.75284 -75.372670 0.00000000
0.00000000 2979047.5
3 0.00000000 4.0 0.00000000 0.5 -.15970769E+09 -2.0
SiO2(cristobalite)
4 3 0.0 2.0 1.0
535.15000 -926547.67 571.80290 -83.513600 -.10104998E-01
0.20917698E-04 1227680.0
3 -.16237635E-07 4.0 -1498.7720 0.5 -46678699. -2.0
1995.9900 -924997.14 566.99971 -83.513600 0.00000000
0.00000000 1227680.0
3 0.00000000 4.0 -1498.7720 0.5 -46678699. -2.0
3000.0000 -961789.83 569.34943 -85.772000 0.00000000
0.00000000 0.00000000
3 0.00000000 4.0 0.00000000 0.5 0.00000000 -2.0
SiO2(liquid)
4 2 0.0 2.0 1.0
1995.9900 -915415.78 562.19940 -83.513600 0.00000000
0.00000000 1227680.0
3 0.00000000 4.0 -1498.7720 0.5 -46678699. -2.0
3000.0000 -952208.47 564.54913 -85.772000 0.00000000
0.00000000 0.00000000
3 0.00000000 4.0 0.00000000 0.5 0.00000000 -2.0
#############################################################################
Date : Nov.1992
Originator : KH+GE
Serial No : COSI.DAT
Quality Status : A
ChemSage Vers. : 4.0
Temperatures : RT to 3500K
Compositions : All
Applications : Phase equilibria and thermodynamic properties.
This files serves as an example of input for several type
of Gibbs energy data of pure substances, e.g. simple
G-functions with several temperature ranges (Type 1), H-S-Cp
input with implicit phase transformation (Type 7), extended
G-functions with several temperature ranges (Type 4).
Sources of data
Unary : THERDAS (90Spe); silicates from 85Ber
Binary : None
References : 85Ber Berman,R.G.; Brown,T.H.; Greenwood,H.J.: Atomic
Energy of Canada Limited, TR-377, 1985
90Spe Spencer,P.J.; Hack,K. : Swiss Materials 2,3a,
1990, p69
###########################################################################
14.2 CNO.DAT
Directory
Contents of data-file cno.dat System components: 1: C 2: N 3: O Phases: 1: GAS 2: C_DIAMOND_A4 3: C Phase constituents of mixture phases: Phase: GAS Model: IDMX Status: ENTERED 1: C 2: C2 3: C3 4: C4 5: C5 6: CN 7: CN2_CNN 8: CN2_NCN 9: C2N_CCN 10: C2N_CNC 11: CNO 12: CNO_NCO 13: CO 14: CO2 15: C2O 16: N 17: N2 18: N3 19: NO 20: NO2 21: NO3 22: N2O 23: N2O3 24: N2O4 25: N2O5 26: O 27: O2 28: O3
Data-file
C-N-O See commentary below ! GTT 16/09/97
3 1 28 2
C N O
12.0110 14.0067 15.9994
6 1 2 3 4 5 6
4 1 2 3 4
GAS
IDMX
C
7 3 1.0 0.0 0.0
716680.00 158.09900
3400.0000 20.975300 -.39964699E-03 0.20077000E-06 -3361.3000
0.00000000
10000.000 23.050699 0.12092100E-03 -.40457500E-08 -17116500.
0.00000000
20000.000 19.445299 0.30792100E-03 0.36924100E-09 0.11237700E+09
C2
7 4 2.0 0.0 0.0
830457.00 197.09500
500.00000 97.481400 -.16406000 0.11841400E-03 -1381500.0
0.00000000
4300.0000 30.083500 0.52427799E-02 -.49857601E-06 1737000.0
0.00000000
12000.000 27.969601 0.38818000E-02 -.19335900E-06 44760100.
0.00000000
20000.000 86.666603 -.31285000E-02 0.43685599E-07 -.12092200E+10
C3
7 5 3.0 0.0 0.0
839958.00 237.61000
1000.0000 32.215599 0.29097101E-01 -.10668400E-04 200555.00
0.00000000
2800.0000 59.939800 -.50827898E-02 0.15248399E-05 -5538910.0
0.00000000
5100.0000 -7.2863898 0.20966399E-01 -.12041700E-05 0.11744000E+09
0.00000000
8000.0000 60.731098 0.95443502E-02 -.83080499E-06 -.38919699E+09
0.00000000
10000.000 211.87399 -.18404599E-01 0.61829900E-06 -.16880399E+10
C4
7 3 4.0 0.0 0.0
1033920.0 252.85600
2500.0000 53.395401 0.27995300E-01 -.49561600E-05 -362017.00
0.00000000
7000.0000 106.17400 -.33283301E-02 0.24180900E-06 -43813500.
0.00000000
10000.000 92.451797 0.53776300E-03 -.42438199E-07 -15174300.
C5
7 3 5.0 0.0 0.0
1050940.0 271.67200
900.00000 62.971901 0.63403003E-01 -.24581601E-04 -372551.00
0.00000000
3500.0000 106.59700 0.29567401E-02 -.42434201E-06 -7495690.0
0.00000000
10000.000 112.17100 0.13222300E-04 -.66332700E-09 -13131600.
CN
7 6 1.0 1.0 0.0
440287.00 202.64301
1000.0000 23.311701 0.13630500E-01 -.36727999E-05 188321.00
0.00000000
2800.0000 32.865898 0.13884200E-02 0.58599801E-06 -1381760.0
0.00000000
5500.0000 35.624802 0.45816898E-02 -.44317400E-06 -29864200.
0.00000000
9800.0000 49.662998 -.10938901E-02 0.10360400E-06 -10523900.
0.00000000
19000.000 26.034599 0.32982901E-02 -.10904700E-06 86173200.
0.00000000
20000.000 70.670502 -.40738899E-03 -.23514801E-07 -.17567300E+10
CN2_CNN
7 2 1.0 2.0 0.0
633484.00 232.08701
1500.0000 37.080898 0.24793999E-01 -.59896302E-05 -145473.00
0.00000000
6000.0000 69.647003 -.18004900E-02 0.13017301E-06 -14647200.
CN2_NCN
7 3 1.0 2.0 0.0
500535.00 226.22900
700.00000 22.682100 0.74644603E-01 -.37100399E-04 68821.203
0.00000000
1900.0000 65.881699 -.16084200E-03 -.35964300E-06 -4262720.0
0.00000000
6000.0000 64.095901 -.50357298E-03 0.41227899E-07 -687544.00
C2N_CCN
7 3 2.0 1.0 0.0
804806.00 239.02901
900.00000 36.285400 0.36251500E-01 -.15046300E-04 -105900.00
0.00000000
5300.0000 59.165699 0.13928399E-02 -.79184099E-08 -3093670.0
0.00000000
6000.0000 54.098099 0.31922900E-02 -.18162299E-06 8301570.0
C2N_CNC
7 3 2.0 1.0 0.0
684924.00 233.81900
800.00000 35.192699 0.41986100E-01 -.19222200E-04 -85784.703
0.00000000
4000.0000 60.762402 0.80248498E-03 -.10756100E-06 -3195470.0
0.00000000
6000.0000 62.406399 -.13479100E-04 0.10161200E-08 -5052510.0
CNO
7 3 1.0 1.0 1.0
177496.00 232.38100
900.00000 30.857500 0.41810598E-01 -.16743699E-04 -154913.00
0.00000000
3300.0000 58.688599 0.19923300E-02 -.29848300E-06 -4462280.0
0.00000000
6000.0000 62.265900 0.21348900E-04 -.14226700E-08 -7807020.0
CNO_NCO
7 3 1.0 1.0 1.0
159410.00 232.22099
800.00000 28.862801 0.47146101E-01 -.20431900E-04 -92762.602
0.00000000
2000.0000 53.999100 0.63656601E-02 -.13898500E-05 -3099870.0
0.00000000
6000.0000 63.381302 -.60019898E-03 0.92913197E-07 -8626740.0
CO
7 3 1.0 0.0 1.0
-110529.00 197.66499
800.00000 25.846201 0.65631098E-02 0.98167504E-06 111208.00
0.00000000
2200.0000 29.936600 0.54106200E-02 -.10533500E-05 -1082960.0
0.00000000
6000.0000 37.175900 0.20404700E-03 0.38694901E-08 -5448030.0
CO2
7 4 1.0 0.0 2.0
-393510.00 213.78300
900.00000 29.329599 0.39927199E-01 -.14750300E-04 -248862.00
0.00000000
2700.0000 54.438499 0.51133898E-02 -.80510500E-06 -4358740.0
0.00000000
7600.0000 76.003403 -.52149598E-02 0.64008100E-06 -35080900.
0.00000000
10000.000 -85.621300 0.24522400E-01 -.90525100E-06 0.14018900E+10
C2O
7 3 2.0 0.0 1.0
291043.00 233.60800
1000.0000 37.785702 0.29073700E-01 -.94620900E-05 -225213.00
0.00000000
3500.0000 56.629601 0.52282899E-02 -.59135601E-06 -4096730.0
0.00000000
6000.0000 68.436699 0.97912597E-03 -.14914700E-06 -32900900.
N
7 2 0.0 1.0 0.0
472687.00 153.29800
2950.0000 20.893900 -.16910399E-03 0.60112100E-07 -5577.5698
0.00000000
6000.0000 16.376101 0.45674801E-03 0.16739800E-06 15118200.
N2
7 3 0.0 2.0 0.0
0.00000000 191.61099
800.00000 27.223301 0.25198299E-02 0.32362900E-05 76653.398
0.00000000
2200.0000 28.423800 0.63785501E-02 -.12398300E-05 -833938.00
0.00000000
6000.0000 37.550098 0.12318000E-04 0.25352801E-07 -6855020.0
N3
7 3 0.0 3.0 0.0
436000.00 223.06599
800.00000 24.401800 0.50211601E-01 -.20478799E-04 -123306.00
0.00000000
2200.0000 52.349899 0.71044699E-02 -.14389200E-05 -3738980.0
0.00000000
6000.0000 61.964901 0.11107600E-03 -.88627798E-08 -9309660.0
NO
7 5 0.0 1.0 1.0
91277.000 210.74500
1300.0000 23.051201 0.15473500E-01 -.47752401E-05 233303.00
0.00000000
5500.0000 37.556301 -.51918800E-04 0.60612400E-07 -3986150.0
0.00000000
9800.0000 13.212000 0.37948100E-02 -.64632999E-07 0.20715299E+09
0.00000000
14500.000 4.4607501 0.72746398E-02 -.28647401E-06 -.18147400E+09
0.00000000
20000.000 170.93600 -.86434204E-02 0.14216199E-06 -.56028498E+10
NO2
7 3 0.0 1.0 2.0
34193.000 240.16600
800.00000 22.650000 0.52844901E-01 -.22859200E-04 70594.102
0.00000000
2400.0000 54.239700 0.28189200E-02 -.15991800E-06 -3832450.0
0.00000000
6000.0000 46.444000 0.47797300E-02 -.96086502E-07 11854400.
NO3
7 3 0.0 1.0 3.0
71128.000 252.62199
600.00000 13.982300 0.14200200 -.87225097E-04 -143564.00
0.00000000
1300.0000 69.838501 0.14972700E-01 -.48511401E-05 -3489250.0
0.00000000
6000.0000 82.856201 0.95063398E-04 -.83041503E-08 -6635110.0
N2O
7 3 0.0 2.0 1.0
81600.000 220.00500
900.00000 31.214800 0.38315602E-01 -.14092900E-04 -246006.00
0.00000000
2900.0000 55.975101 0.42720600E-02 -.57106701E-06 -4357220.0
0.00000000
6000.0000 62.137299 0.62846800E-03 0.26556201E-07 -9580270.0
N2O3
7 3 0.0 2.0 3.0
86631.000 314.73401
900.00000 56.690399 0.62969401E-01 -.25171499E-04 -45424.500
0.00000000
3600.0000 100.38500 0.13410100E-02 -.55178301E-07 -6993170.0
0.00000000
6000.0000 93.545403 0.34843700E-02 -.22545800E-06 10275000.
N2O4
7 3 0.0 2.0 4.0
11110.000 304.44501
700.00000 54.510300 0.12472000 -.61755600E-04 -627855.00
0.00000000
2400.0000 121.09400 0.57820301E-02 -.11838400E-05 -7002920.0
0.00000000
6000.0000 128.78999 0.22902101E-04 -.17693800E-08 -10930000.
N2O5
7 3 0.0 2.0 5.0
13300.000 355.71701
800.00000 76.767700 0.10727900 -.47430101E-04 -820876.00
0.00000000
3000.0000 141.33299 0.50176601E-02 -.83227502E-06 -8874420.0
0.00000000
6000.0000 149.48500 0.40587100E-04 -.27888700E-08 -15036600.
O
7 2 0.0 0.0 1.0
249174.00 161.06000
2950.0000 21.015600 -.25374099E-03 0.73878802E-07 85794.203
0.00000000
6000.0000 17.211901 0.10827100E-02 -.45871200E-07 7946340.0
O2
7 5 0.0 0.0 2.0
0.00000000 205.14700
900.00000 22.258600 0.20477301E-01 -.80396803E-05 153499.00
0.00000000
3700.0000 33.557301 0.24697999E-02 -.10016600E-06 -1079770.0
0.00000000
9600.0000 24.479799 0.52695200E-02 -.36092700E-06 30241900.
0.00000000
18500.000 87.561401 -.51574502E-02 0.11272600E-06 -.58104998E+09
0.00000000
20000.000 63.737000 -.28750000E-02 0.54000001E-07 -.50307798
O3
7 7 0.0 0.0 3.0
141800.00 239.00500
700.00000 27.581200 0.55261500E-01 -.27632401E-04 -199061.00
0.00000000
1300.0000 60.102901 -.41291201E-02 0.31049201E-05 -3144350.0
0.00000000
2100.0000 134.37000 -.79414703E-01 0.24627499E-04 -24724500.
0.00000000
2800.0000 -421.20001 0.25682199 -.32686101E-04 0.42609699E+09
0.00000000
3500.0000 -223.44400 0.18447199 -.25841100E-04 43179700.
0.00000000
4900.0000 764.84302 -.19705500 0.15587100E-04 -.19221700E+10
0.00000000
6000.0000 149.96100 -.25671501E-01 0.21330600E-05 0.43400000E+09
C_DIAMOND_A4
4 1 1.0 0.0 0.0
6000.0000 -16359.441 175.61000 -24.310000 -.47230000E-03
0.00000000 2698000.0
2 -.26100000E+09 -2.0 0.11100000E+11 -3.0
C
7 3 1.0 0.0 0.0
0.00000000 5.6859999
800.00000 -3.0257599 0.47484200E-01 -.23528601E-04 -45617.301
0.00000000
1900.0000 20.021799 0.48779999E-02 -.11859501E-05 -2133160.0
0.00000000
6000.0000 25.499001 -.19641699E-03 0.95123198E-07 -3795520.0
#############################################################################
Date : 16.09.1997
Originator : KH
Serial No : CNO.DAT
Quality Status : A
ChemSage Vers. : 4.0
Temperatures : All
Compositions : All
Applications : Combustion equilibria and thermodynamic properties of
the gas species and condensed phases.
Sources of data
Unary : 96SGT and 91Din
Binary : None
References : 91Din Dinsdale,A.T. CALPHAD 15(4), 1991, 317-427
96SGT SGTE Pure Substance database, Edition 1996
###########################################################################
14.3 FEC.DAT
Directory
Contents of data-file fec.dat System components: 1: Fe 2: C Phases: 1: GAS 2: LIQUID 3: BCC_A2 4: FCC_A1 5: C_GRAPHITE 6: Fe3C_CEMENTITE Phase constituents of mixture phases: Phase: GAS Model: IDMX Status: ENTERED 1: C 2: C2 3: C3 4: C4 5: C5 6: C6 7: C7 8: Fe Phase: LIQUID Model: RKMP Status: ENTERED 1: C 2: Fe Phase: BCC_A2 Model: SUBLM Status: ENTERED 1: Fe:C 2: Fe:Va Phase: FCC_A1 Model: RKMPM Status: ENTERED 1: FeC 2: Fe
Data-file
Fe-C See commentary below ! GTT 09/11/98 SSL92N05
2 4 8 2 2 2 2
Fe C
55.8470 12.0110
6 1 2 3 4 5 6
4 1 2 3 4
GAS
IDMX
C
4 2 0.0 1.0
3000.0000 710400.00 -17.077222 -21.054000 0.25309000E-03
-.38666700E-07 3707.6000
1 0.00000000 0.0
6000.0000 713129.05 -38.618592 -18.170000 -.73850000E-03
0.17796700E-07 75203.000
1 0.00000000 0.0
C2
4 3 0.0 2.0
400.00000 809200.49 474.13666 -92.200716 0.86155380E-01
-.23101541E-04 476557.60
1 0.00000000 0.0
1600.0000 828935.55 82.995371 -28.598477 -.36201851E-02
0.23011303E-06 -657802.20
1 0.00000000 0.0
6000.0000 820236.64 137.47883 -35.901230 -.72968960E-03
0.10299613E-08 1372216.0
1 0.00000000 0.0
C3
4 3 0.0 3.0
1200.0000 833687.45 97.202195 -35.052715 -.12125148E-01
0.12964236E-05 -59322.844
1 0.00000000 0.0
3800.0000 807138.57 322.40377 -66.622669 0.47850316E-02
-.42562298E-06 4179196.8
1 0.00000000 0.0
6000.0000 1044778.6 -608.06348 48.889622 -.19275960E-01
0.49025880E-06 -82501748.
1 0.00000000 0.0
C4
4 3 0.0 4.0
1200.0000 1003706.3 204.70568 -52.057328 -.18956030E-01
0.18511236E-05 199317.39
1 0.00000000 0.0
4400.0000 973125.87 452.52662 -86.585370 -.10813130E-02
0.51440188E-07 5399339.0
1 0.00000000 0.0
6000.0000 761565.21 1007.2673 -152.03819 0.81392607E-02
-.19964026E-06 0.13586479E+09
1 0.00000000 0.0
C5
4 4 0.0 5.0
1000.0000 1020031.2 278.54107 -64.503473 -.26696066E-01
0.29275580E-05 299459.34
1 0.00000000 0.0
2200.0000 993156.23 570.71476 -107.07986 0.27493482E-02
-.82689020E-06 3417001.7
1 0.00000000 0.0
3800.0000 1142512.8 -294.09095 6.7835192 -.35082756E-01
0.14932061E-05 -32394323.
1 0.00000000 0.0
6000.0000 445025.44 2102.1813 -286.10485 0.19510557E-01
-.41218955E-06 0.27869886E+09
1 0.00000000 0.0
C6
4 3 0.0 6.0
1000.0000 1246184.9 311.98451 -74.835861 -.37038337E-01
0.40882770E-05 241241.07
1 0.00000000 0.0
2800.0000 1200861.1 731.55484 -134.46246 -.19671766E-02
0.18033180E-06 6717468.5
1 0.00000000 0.0
6000.0000 1214236.9 736.70494 -136.25991 0.66676224E-03
-.38766155E-07 -3611507.5
1 0.00000000 0.0
C7
4 4 0.0 7.0
1000.0000 1317471.1 445.39667 -98.716041 -.31425961E-01
0.26642952E-05 404461.00
1 0.00000000 0.0
2000.0000 1351184.3 237.07793 -71.712505 -.36116999E-01
0.18958680E-05 -6064281.2
1 0.00000000 0.0
3600.0000 1036356.3 1692.1807 -256.70179 0.99385272E-02
-.10190411E-06 91042911.
1 0.00000000 0.0
6000.0000 1158858.1 1431.4995 -227.61127 0.88048096E-02
-.21646063E-06 13357757.
1 0.00000000 0.0
Fe
4 3 1.0 0.0
950.00000 406417.00 36.785878 -33.007900 0.92758000E-02
-.13799600E-05 112882.00
1 0.00000000 0.0
2350.0000 415082.70 -53.396222 -19.902900 0.13565300E-03
-.13883100E-06 -996892.00
1 0.00000000 0.0
6000.0000 407130.10 -32.854222 -22.159400 -.14191800E-03
-.68989700E-07 2445910.0
1 0.00000000 0.0
LIQUID
RKMP
C
4 1 0.0 1.0
6000.0000 100000.56 146.10000 -24.300000 -.47230000E-03
0.00000000 2562600.0
2 -.26430000E+09 -2.0 0.12000000E+11 -3.0
Fe
4 2 1.0 0.0
1811.0000 13265.870 117.57557 -23.514300 -.43975200E-02
-.58927000E-07 77359.000
1 -.36751551E-20 7.0
6000.0000 -10838.830 291.30200 -46.000000 0.00000000
0.00000000 0.00000000
1 0.00000000 0.0
2
1 2 3
-124320.00 28.500000 0.00000000 0.00000000
19300.000 0.00000000 0.00000000 0.00000000
49260.000 -19.000000 0.00000000 0.00000000
0
BCC_A2
SUBLM
1.00000 0.400000
Fe:C
16 2 1.0 3.0
1811.0000 271170.38 711.99100 -96.414300 -.58144200E-02
-.58927000E-07 7765159.0
2 -.79290000E+09 -2.0 0.36000000E+11 -3.0
6000.0000 244561.10 887.16955 -118.90000 -.14169000E-02
0.00000000 7687800.0
3 0.22960300E+32 -9.0 -.79290000E+09 -2.0 0.36000000E+11 -3.0
1043.00 2.22000
Fe:Va
16 2 1.0 0.0
1811.0000 1225.7000 124.13400 -23.514300 -.43975200E-02
-.58927000E-07 77359.000
1 0.00000000 0.0
6000.0000 -25383.581 299.31255 -46.000000 0.00000000
0.00000000 0.00000000
1 0.22960300E+32 -9.0
1043.00 2.22000
2
1.00000 3.00000
1 2
Fe
C Va
1 1
1 2
0
3
1 2 3 1
0.00000000 -190.00000 0.00000000 0.00000000
0
FCC_A1
RKMPM
0.333333 0.280000
FeC
16 2 1.0 1.0
1811.0000 59601.859 287.26900 -48.964300 -.42298200E-02
-.58927000E-07 2639959.0
2 -.26430000E+09 -2.0 0.12000000E+11 -3.0
6000.0000 32741.163 455.10556 -70.300000 -.47230000E-03
0.00000000 2562600.0
3 0.27885400E+32 -9.0 -.26430000E+09 -2.0 0.12000000E+11 -3.0
-201.000 -2.10000
Fe
16 2 1.0 0.0
1811.0000 -236.70000 132.41600 -24.664300 -.37575200E-02
-.58927000E-07 77359.000
1 0.00000000 0.0
6000.0000 -27097.396 300.25256 -46.000000 0.00000000
0.00000000 0.00000000
1 0.27885400E+32 -9.0
-201.000 -2.10000
0
2
1 2 1
-34671.000 0.00000000 0.00000000 0.00000000
0
C_GRAPHITE
4 1 0.0 1.0
6000.0000 -17368.441 170.73000 -24.300000 -.47230000E-03
0.00000000 2562600.0
2 -.26430000E+09 -2.0 0.12000000E+11 -3.0
Fe3C_CEMENTITE
4 1 3.0 1.0
6000.0000 -10745.000 706.04000 -120.60000 0.00000000
0.00000000 0.00000000
1 0.00000000 0.0
#############################################################################
Date : 12.11.1998
Originator : KH
Serial No : FEC.DAT
Quality Status : A
ChemSage Vers. : 4.0
Temperatures : RT to 2500K
Compositions : All
Applications : Phase equilibria; phase diagram, also metastable with
CEMENTITE; thermodynamic properties of the phases and
their constituents as well as the system components.
Sources of data
Unary : 91Din
Binary : 85Gus
Species and interaction list:
GAS NOTE: The GAS phase is treated as an ideal mixture.
IDMX However, the speciation of the carbon is treated
C such that the calculated vapour pressures are
C2 for a proper gas.
C3
C4
C5
C6
C7
Fe
LIQUID NOTE: The LIQUID phase is treated as a simple
RKMP substitutional solution with the elements
C as phase constituents.
Fe
Interaction term is available for:
C-Fe
BCC_A2 NOTE: The BCC_A2 phase is described with a two-
SUBLM sublattice model, the metal sublattice
Fe:C being occupied by Fe only, and the inter-
Fe:Va stitial site by C or vacancies.
Sublattice occupancy:
Metal Sublattice: Fe
Interstitial sites: C, Va
Site ratio is: (Fe)1(C,Va)3
Interaction term is available for:
Fe:C,Va This is a non-ideal term for interaction between
carbon and vacancies in the presence of iron.
FCC_A1 NOTE: The representation of FCC_A1 as an
RKMPM Associated Solution with FeC and Fe
FeC as constituents is mathematically
Fe exactly equal to the Sublattice Description
FeC-Fe that is used in higher order systems.
The ChemAccess software however recognizes
that (Fe)1(C,Va)1 is mathematically the
same as FeC-Fe and therefore uses the
simple RKMPM (Redlich-Kister with Magnetic
Terms) formalism.
Stoichiometric Pure Substances
C_GRAPHITE NOTE: Carbon as GRAPHITE and Fe3C (Cementite) are
Fe3C_CEMENTITE the stoichiometric solid phases in this system.
In order to calculate metastable equilibria
involving Fe3C the C_GRAPHITE phase must be
set DORMANT.
References : 85Gus Gustafson,P. Scan.J.Metall. Vol 14, 1985, p259-267
91Din Dinsdale,A.T. CALPHAD, Vol 15(4), 1991, p317-427
###########################################################################
14.4 FEMGSIO4.DAT
Directory
Contents of data-file femgsio4.dat System components: 1: Fe2SiO4 2: Mg2SiO4 Phases: 1: Olivine 2: Spinell 3: beta-phase 4: Fe2SiO4 5: Mg2SiO4 Phase constituents of mixture phases: Phase: Olivine Model: RKMP Status: ENTERED 1: FeSi0.5O2 2: MgSi0.5O2 Phase: Spinell Model: RKMP Status: ENTERED 1: FeSi0.5O2 2: MgSi0.5O2 Phase: beta-phase Model: RKMP Status: ENTERED 1: FeSi0.5O2 2: MgSi0.5O2
Data-file
Fe2SiO4-Mg2SiO4, See commentary below ! GTT 16.8.96
2 4 0 2 2 2 2
Fe2SiO4 Mg2SiO4
203.7778 140.7076
6 1 2 3 4 5 6
1 1
Olivine
RKMP
FeSi0.5O2
12 1 .5000 0.0
-739085.00 317.33200
2000.0000 127.66500 0.00000000 0.00000000 0.00000000
3 -1061.7000 -0.5 83150000. -3.0 0.00000000 0.0
23.1400 0.266000E-04 0.873600E-08 0.000000 -.248700 0.725160
0.000000 0.000000 0.000000 4.00000 0.000000
MgSi0.5O2
12 1 0.0 .5000
-1088600.0 294.28300
2000.0000 108.89700 0.00000000 0.00000000 0.00000000
3 -704.10000 -0.5 -.26318000E+09 -3.0 0.00000000 0.0
21.8350 0.305200E-04 0.850400E-08 0.000000 -.582400 0.773990
0.000000 0.000000 0.000000 4.88000 0.000000
2
1 2 1
-8314.0000
0
Spinell
RKMP
FeSi0.5O2
12 1 .5000 0.0
-738334.00 310.27300
2000.0000 130.98600 0.00000000 0.00000000 0.00000000
3 -1201.4000 -0.5 0.13419000E+09 -3.0 0.00000000 0.0
21.0100 0.245500E-04 0.359100E-08 0.000000 -.370300 0.507610
0.000000 0.000000 0.000000 4.00000 0.000000
MgSi0.5O2
12 1 0.0 .5000
-1069300.0 289.97300
2000.0000 100.09600 0.00000000 0.00000000 -660000.00
2 -518.40000 -0.5 -.24582000E+09 -3.0
19.7650 0.236700E-04 0.529800E-08 0.000000 -.570200 0.469480
0.000000 0.000000 0.000000 4.00000 0.000000
2
1 2 1
-9977.0000
0
beta-phase
RKMP
FeSi0.5O2
12 1 .5000 0.0
-736473.00 312.76200
2000.0000 136.33200 0.00000000 0.00000000 0.00000000
3 -1319.8000 -0.5 0.22412000E+09 -3.0 0.00000000 0.0
21.5750 0.231900E-04 0.711700E-08 0.000000 -.243000 0.602410
0.000000 0.000000 0.000000 4.00000 0.000000
MgSi0.5O2
12 1 0.0 .5000
-1072450.0 291.75300
2000.0000 108.21800 0.00000000 0.00000000 0.00000000
3 -736.20000 -0.5 -.27895000E+09 -3.0 0.00000000 0.0
20.2700 0.271100E-04 0.688500E-08 0.000000 -.576700 0.625000
0.000000 0.000000 0.000000 4.00000 0.000000
2
1 2 1
-11210.000
0
Fe2SiO4
1 1 1.0 0.0
2500.0000 0.00000000 0.00000000 0.00000000 0.00000000
0.00000000 0.00000000
Mg2SiO4
1 1 0.0 1.0
2500.0000 0.00000000 0.00000000 0.00000000 0.00000000
0.00000000 0.00000000
#############################################################################
Date : 16.8.1996
Originator : KH
Serial No : femgsio4.dat
Quality Status : A
ChemSage Vers. : 4.0
Temperatures : Below liquidus
Compositions : All
Applications : Solid T-P-composition equilibria and thermal properties in
the system Fe2SiO4 - Mg2SiO4 for pressures between 1 bar and
300 kbar.
NOTE: The system is Fe2SiO4 - Mg2SiO4. However, in order
to obtain the correct ideal entropy between Fe and Mg
the scaling factor 0.5 has been applied for the phase
constituents. The two Stoichiometric Condensed phases
Fe2SiO4 and Mg2SiO4 do not take part in any equilibrium
but serve as simple input substances for the definition
of the system composition.
Sources of data
Unary : 86Fei
Binary : 86Fei
Species and interaction list:
Olivine
FeSi0.5O2
MgSi0.5O2
Spinell
FeSi0.5O2
MgSi0.5O2
beta-phase
FeSi0.5O2
MgSi0.5O2
References : 86Fei Yingwei Fei; Surendra Saxena:
Chem Minerals 13, 1986, pp311-324
###########################################################################
14.5 PBSN.DAT
Directory
Contents of data-file pbsn.dat System components: 1: Pb 2: Sn Phases: 1: LIQUID 2: BCT_A5#1 3: BCT_A5#2 4: FCC_A1 Phase constituents of mixture phases: Phase: LIQUID Model: RKMP Status: ENTERED 1: Pb 2: Sn Phase: BCT_A5#1 Model: RKMP Status: ENTERED 1: Pb 2: Sn Phase: BCT_A5#2 Model: RKMP Status: ENTERED 1: Pb 2: Sn Phase: FCC_A1 Model: RKMP Status: ENTERED 1: Pb 2: Sn
Data-file
Pb-Sn See commentary below ! GTT 22-2-1994
2 5 0 2 2 2 2 0
Pb Sn
207.2000 118.7100
6 1 2 3 4 5 6
2 1 2
LIQUID
RKMP
Pb
4 3 1.0 0.0
600.65000 -2977.9280 93.964931 -24.524223 -.36589500E-02
-.24395000E-06 0.00000000
3 0.00000000 -9.0 -.60144000E-18 7.0 0.00000000 0.0
1200.0000 -5678.0030 146.19157 -32.491396 0.15461300E-02
0.00000000 0.00000000
3 0.00000000 -9.0 0.00000000 7.0 0.00000000 0.0
5000.0000 9010.7080 45.087458 -18.964064 -.28829430E-02
0.98144000E-07 -2696755.0
3 0.00000000 -9.0 0.00000000 7.0 0.00000000 0.0
Sn
4 3 0.0 1.0
505.08000 1249.0870 51.337011 -15.961000 -.18870200E-01
0.31211670E-05 -61960.000
3 0.00000000 -9.0 0.14931665E-17 7.0 0.00000000 0.0
800.00000 9495.4290 -9.8234570 -8.2590490 -.16814429E-01
0.26231310E-05 -1081244.0
3 0.18420000E+24 -9.0 0.00000000 7.0 0.00000000 0.0
3000.0000 -1286.2540 125.16815 -28.451200 0.00000000
0.00000000 0.00000000
3 0.18420000E+24 -9.0 0.00000000 7.0 0.00000000 0.0
2
1 2 2
5125.0000 1.4642400
293.82000 0.00000000
0
BCT_A5
RKMP
Pb
4 3 1.0 0.0
600.65000 -7161.0850 105.23519 -24.524223 -.36589500E-02
-.24395000E-06 0.00000000
3 0.00000000 -9.0 0.00000000 7.0 0.00000000 0.0
1200.0000 -10042.115 157.77815 -32.491396 0.15461300E-02
0.00000000 0.00000000
3 0.80564400E+26 -9.0 0.00000000 7.0 0.00000000 0.0
5000.0000 4646.5960 56.674045 -18.964064 -.28829430E-02
0.98144000E-07 -2696755.0
3 0.80564400E+26 -9.0 0.00000000 7.0 0.00000000 0.0
Sn
4 4 0.0 1.0
250.00000 -7958.5170 122.75003 -25.858000 0.51185000E-03
-.31927670E-05 18440.000
3 0.00000000 -9.0 0.00000000 7.0 0.00000000 0.0
505.08000 -5855.1350 65.427891 -15.961000 -.18870200E-01
0.31211670E-05 -61960.000
3 0.00000000 -9.0 0.00000000 7.0 0.00000000 0.0
800.00000 2524.7240 3.9898450 -8.2590490 -.16814429E-01
0.26231310E-05 -1081244.0
3 -.12307000E+26 -9.0 0.00000000 7.0 0.00000000 0.0
3000.0000 -8256.9590 138.98146 -28.451200 0.00000000
0.00000000 0.00000000
3 -.12307000E+26 -9.0 0.00000000 7.0 0.00000000 0.0
2
1 2 1
17117.780 -11.806560
0
BCT_A5
RKMP
Pb
4 3 1.0 0.0
600.65000 -7161.0850 105.23519 -24.524223 -.36589500E-02
-.24395000E-06 0.00000000
3 0.00000000 -9.0 0.00000000 7.0 0.00000000 0.0
1200.0000 -10042.115 157.77815 -32.491396 0.15461300E-02
0.00000000 0.00000000
3 0.80564400E+26 -9.0 0.00000000 7.0 0.00000000 0.0
5000.0000 4646.5960 56.674045 -18.964064 -.28829430E-02
0.98144000E-07 -2696755.0
3 0.80564400E+26 -9.0 0.00000000 7.0 0.00000000 0.0
Sn
4 4 0.0 1.0
250.00000 -7958.5170 122.75003 -25.858000 0.51185000E-03
-.31927670E-05 18440.000
3 0.00000000 -9.0 0.00000000 7.0 0.00000000 0.0
505.08000 -5855.1350 65.427891 -15.961000 -.18870200E-01
0.31211670E-05 -61960.000
3 0.00000000 -9.0 0.00000000 7.0 0.00000000 0.0
800.00000 2524.7240 3.9898450 -8.2590490 -.16814429E-01
0.26231310E-05 -1081244.0
3 -.12307000E+26 -9.0 0.00000000 7.0 0.00000000 0.0
3000.0000 -8256.9590 138.98146 -28.451200 0.00000000
0.00000000 0.00000000
3 -.12307000E+26 -9.0 0.00000000 7.0 0.00000000 0.0
2
1 2 1
17117.780 -11.806560
0
FCC_A1
RKMP
Pb
4 3 1.0 0.0
600.65000 -7650.0850 101.71519 -24.524223 -.36589500E-02
-.24395000E-06 0.00000000
3 0.00000000 -9.0 0.00000000 7.0 0.00000000 0.0
1200.0000 -10531.115 154.25815 -32.491396 0.15461300E-02
0.00000000 0.00000000
3 0.80564400E+26 -9.0 0.00000000 7.0 0.00000000 0.0
5000.0000 4157.5960 53.154045 -18.964064 -.28829430E-02
0.98144000E-07 -2696755.0
3 0.80564400E+26 -9.0 0.00000000 7.0 0.00000000 0.0
Sn
4 3 0.0 1.0
505.08000 -1705.1350 60.227891 -15.961000 -.18870200E-01
0.31211670E-05 -61960.000
3 0.00000000 -9.0 0.00000000 7.0 0.00000000 0.0
800.00000 6674.7240 -1.2101550 -8.2590490 -.16814429E-01
0.26231310E-05 -1081244.0
3 -.12307000E+26 -9.0 0.00000000 7.0 0.00000000 0.0
3000.0000 -4106.9590 133.78146 -28.451200 0.00000000
0.00000000 0.00000000
3 -.12307000E+26 -9.0 0.00000000 7.0 0.00000000 0.0
2
1 2 1
5132.4100 1.5631200
0
#############################################################################
Date : 22.2.94
Originator : KH
Serial No : PBSN.DAT
Quality Status : A
ChemSage Vers. : 4.0
Temperatures : RT - 1500 K
Compositions : All
Applications : Phase equilibria and all thermodynamic properties
NOTE: The double entry of the BCT_A5 phase (Sn-terminal
solution) is required because of the inherent meta-
stable miscibility gap.
Sources of data
Unary : 92SGT
Binary : 92SGT
Species and interaction list:
LIQUID
Pb
Sn
Pb-Sn
BCT_A5
Pb
Sn
Pb-Sn
FCC_A1
Pb
Sn
Pb-Sn
References : 92SGT SGTE Solution database, Edition 1992
###########################################################################
14.6 PITZER.DAT
Directory
Contents of data-file pitzer.dat System components: 1: Na 2: Cl 3: C 4: O 5: H 6: EA Phases: 1: GAS 2: AQUEOUS 3: NaHCO3 4: Na2CO3.10H2O 5: Na2CO3.7H2O 6: Na2CO3.H2O 7: Na3H(CO3)2(H2O)2 8: NaCl Phase constituents of mixture phases: Phase: GAS Model: IDMX Status: ENTERED 1: CO 2: CO2 3: H2 4: H2O 5: O2 Phase: AQUEOUS Model: PITZ Status: ENTERED 1: H2O 2: H<+> 3: Na<+> 4: OH<-> 5: HCO3<-> 6: CO3<2-> 7: CO2(AQ) 8: Cl<->
Data-file
H<+>-Na<+>-CO3<2->-Cl<->-H2O at 298.15 K See commentary below! GTT Nov.1992
6 2 5 8 6
Na Cl C
O H EA
22.9898 35.4530 12.0110
15.9994 1.0080 0.0000
1 1
1 2
GAS
IDMX
CO
1 1 0.0 0.0 1.0 1.0 0.0 0.0
298.15000 -137169.00
CO2
1 1 0.0 0.0 1.0 2.0 0.0 0.0
298.15000 -394384.48
H2
1 1 0.0 0.0 0.0 0.0 2.0 0.0
298.15000 0.00000000
H2O
1 1 0.0 0.0 0.0 1.0 2.0 0.0
298.15000 -228587.00
O2
1 1 0.0 0.0 0.0 2.0 0.0 0.0
298.15000 0.00000000
AQUEOUS
PITZ
H2O
1 1 0.0 0.0 0.0 0.0 1.0 2.0 0.0
298.15000 -237147.06
H<+>
1 1 1.0 0.0 0.0 0.0 0.0 1.0 -1.0
298.15000 0.00000000
Na<+>
1 1 1.0 1.0 0.0 0.0 0.0 0.0 -1.0
298.15000 -261905.78
OH<->
1 1 -1.0 0.0 0.0 0.0 1.0 1.0 1.0
298.15000 -157253.54
HCO3<->
1 1 -1.0 0.0 0.0 1.0 3.0 1.0 1.0
298.15000 -586898.90
CO3<2->
1 1 -2.0 0.0 0.0 1.0 3.0 0.0 2.0
298.15000 -527882.03
CO2(AQ)
1 1 0.0 0.0 0.0 1.0 2.0 0.0 0.0
298.15000 -385926.23
Cl<->
1 1 -1.0 0.0 1.0 0.0 0.0 0.0 1.0
298.15000 -131273.92
2
2 3 0 0.29932236
3
2 3 8 -.33258040E-01
2
2 8 0 1.4758255
2
2 8 1 2.4486232
2
2 8 3 0.66516080E-02
2
3 4 0 0.71837366
2
3 4 1 2.1035710
2
3 4 3 0.36583844E-01
3
3 4 5 -.14134667
3
3 4 8 -.49887060E-01
2
3 5 0 0.23031193
2
3 5 1 0.34172636
3
3 5 6 0.16629020E-01
3
3 5 8 -.12471765
2
3 6 0 0.33174895
2
3 6 1 11.548854
2
3 6 3 0.36583844E-01
3
3 6 8 0.70673335E-01
2
3 7 0 0.83145100
2
3 8 0 0.63606002
2
3 8 1 2.2149855
2
3 8 3 0.10559427E-01
2
4 5 0 0.83145100
2
4 8 0 -.41572550
2
5 6 0 -.33258040
2
5 8 0 0.24943530
2
6 8 0 -.16629020
2
7 8 0 -.41572550E-01
0
NaHCO3
1 1 1.0 0.0 1.0 3.0 1.0 0.0
298.15000 -851105.17
Na2CO3.10H2O
1 1 2.0 0.0 1.0 13.0 20.0 0.0
298.15000 -3427871.7
Na2CO3.7H2O
1 1 2.0 0.0 1.0 10.0 14.0 0.0
298.15000 -2714349.5
Na2CO3.H2O
1 1 2.0 0.0 1.0 4.0 2.0 0.0
298.15000 -1286090.2
Na3H(CO3)2(H2O)2
1 1 3.0 0.0 2.0 8.0 5.0 0.0
298.15000 -2380745.3
NaCl
1 1 1.0 1.0 0.0 0.0 0.0 0.0
298.15000 -384215.74
###########################################################################
Date : Nov.1992
Originator : GE+KH
Serial No : PITZER.DAT
Quality Status : B
ChemSage Vers. : 4.0
Temperatures : 298.15 K
Compositions : Aqueous solutions with up to 10 molality of dissolved
salts
Applications : Natural waters with sodium
This file serves as an example of input for the Pitzer
aqueous model with non-ideal interactions between two
ions of equal or opposite sign and also uncharged species.
Sources of data:
Unary : 84Har
Binary : 84Har
References : 84Har Harvie, C.E.; Moeller, N.; Weare, J.H.: Geochim.
Cosmochim. Acta, 1984, 48, 723
###########################################################################
14.7 WATER.DAT
Directory
Contents of data-file water.dat System components: 1: O 2: H 3: EA Phases: 1: gas_real 2: aqueous 3: H2O_Ice(s) Phase constituents of mixture phases: Phase: gas_real Model: IDVD Status: ENTERED 1: H 2: H2 3: O 4: O2 5: O3 6: OH 7: H2O 8: HOO 9: HOOH Phase: aqueous Model: IDWZ Status: ENTERED 1: H2O_liquid 2: H[+] 3: H2 4: O2 5: OH[-] 6: HO2[-] 7: HOOH
Data-file
System O-H-EA
3 2 9 7 1
O H EA
15.999400 1.007940 0.000549
6 1 2 3 4 5 6
6 1 2 3 4 5 6
gas_real
IDVD
H
4 1 0.0 1.0 0.0
6000.0000 211801.65 24.500245 -20.786000 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
0.00000 0.00000 0.00000 0.00000 0.00000
H2
4 4 0.0 2.0 0.0
1200.0000 -13779.823 -17.824781 -19.825631 -.15388637E-02
-.23830749E-06 147589.95
1 779.44498 0.50
4100.0000 330318.97 -121.44530 -14.387460 -.10320538E-02
0.0000000 -10416577.
2 -74353.404 99.00 8539.3383 0.50
6000.0000 5123954.0 -941.72642 42.957060 0.0000000
0.0000000 -.36536589E+09
2 -931953.90 99.00 68151.779 0.50
6001.0000 -43508.638 176.80798 -41.974318 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
33.2000 13.0000 65.1000 -.218000 0.00000
O
4 3 1.0 0.0 0.0
2200.0000 242682.20 -30.020448 -19.964968 -.13194946E-03
0.61589319E-08 -29327.439
1 83.809856 0.50
6000.0000 -84471.471 186.78944 -38.050669 0.0000000
0.0000000 12120739.
2 70811.436 99.00 -8330.8535 0.50
6001.0000 236685.00 -8.3771889 -22.291487 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
0.00000 0.00000 0.00000 0.00000 0.00000
O2
4 4 2.0 0.0 0.0
1000.0000 -5219.3324 -12.179856 -26.924057 -.84893408E-02
0.11276942E-05 -114664.63
1 -316.64663 0.50
4000.0000 -389938.78 638.67678 -89.681327 0.72372244E-03
0.0000000 9341343.0
2 -16506.149 0.50 95803.960 99.00
6000.0000 -8951197.1 2742.7612 -249.17312 0.0000000
0.0000000 0.59248921E+09
2 -139742.68 0.50 1674792.2 99.00
6001.0000 -42014.302 116.92408 -44.371534 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
154.600 50.4000 73.4000 0.250000E-01 0.00000
O3
4 3 3.0 0.0 0.0
1900.0000 200310.28 315.13014 -76.555764 0.46115697E-02
-.32267167E-06 -821844.45
1 -15871.007 99.00
6000.0000 -95706.532 344.94952 -73.713624 0.0000000
0.0000000 9320171.5
2 47079.583 99.00 -6138.3410 0.50
6001.0000 107048.81 191.83924 -61.231023 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
261.100 55.7000 88.9000 0.691000 0.600000
OH
4 4 1.0 1.0 0.0
1300.0000 15124.539 -53.228376 -20.563267 -.36262413E-02
0.34198989E-07 189112.08
1 3436.3138 99.00
4000.0000 349207.50 -117.24675 -20.415378 -.46215318E-03
0.0000000 -9626333.3
2 -69526.518 99.00 7577.0060 0.50
6000.0000 -1183915.3 523.77324 -73.005362 0.0000000
0.0000000 79896619.
2 230246.83 99.00 -20920.359 0.50
6001.0000 6770.0965 99.135663 -39.420964 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
0.00000 0.00000 0.00000 0.00000 0.00000
H2O
4 4 1.0 2.0 0.0
1100.0000 -255475.81 -15.282587 -25.781640 -.74748581E-02
0.92059316E-07 13999.660
1 1107.2718 99.00
4000.0000 152152.28 164.70757 -53.145789 -.80540038E-04
0.0000000 -12075583.
2 -83128.276 99.00 5947.3700 0.50
6000.0000 -4469439.5 1472.4424 -155.19083 0.0000000
0.0000000 0.29826420E+09
2 778290.99 99.00 -64372.340 0.50
6001.0000 -302989.76 260.62247 -60.574924 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
647.300 221.200 57.1000 0.344000 1.80000
HOO
4 3 2.0 1.0 0.0
2100.0000 15697.283 255.13384 -62.913975 -.11689159E-02
0.0000000 -405639.23
2 -2663.7114 99.00 -1996.0491 0.50
6000.0000 1105560.9 -244.54895 -24.892588 0.0000000
0.0000000 -42897102.
2 -231446.38 99.00 22310.750 0.50
6001.0000 -41569.824 209.48530 -60.708998 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
0.00000 0.00000 0.00000 0.00000 0.00000
HOOH
4 2 2.0 2.0 0.0
1500.0000 -85550.955 331.73213 -78.314851 0.26315727E-02
-.53029881E-06 -685226.23
1 -14710.134 99.00
1501.0000 -166774.40 243.96699 -68.381500 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
0.00000 0.00000 0.00000 0.00000 0.00000
aqueous
IDWZ
H2O_liquid
4 2 0.00 1.0 2.0 0.0
500.00000 -256638.94 -1118.6247 203.11898 -.76034980
0.53188744E-03 -1924378.8
1 -.20591320E-06 4.00
501.00000 -312003.88 493.71232 -83.664491 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
H[+]
4 1 1.00 0.0 1.0 -1.0
500.00000 0.0000000 0.0000000 0.0000000 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
H2
4 2 0.00 0.0 2.0 0.0
573.00000 144981.35 -2046.7532 305.31100 -.34285150
0.0000000 -13210150.
1 0.0000000 0.00
574.00000 -59804.190 1080.3122 -168.06580 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
O2
4 2 0.00 2.0 0.0 0.0
400.00000 -50908.338 967.47122 -162.23000 -.16062500E-01
0.0000000 -1580700.0
1 0.0000000 0.00
401.00000 -69285.338 1192.4835 -194.83875 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
OH[-]
4 2 -1.00 1.0 1.0 1.0
573.00000 -410973.14 3188.5513 -506.37700 0.59067000
0.0000000 12301150.
1 0.0000000 0.00
574.00000 -131167.15 -1698.6943 245.46267 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
HO2[-]
4 2 -1.00 2.0 1.0 1.0
573.00000 -490620.53 5465.2599 -878.65255 1.1414036
0.0000000 25322823.
1 0.0000000 0.00
574.00000 60908.995 -4053.0271 583.64867 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
HOOH
4 2 0.00 2.0 2.0 0.0
473.00000 19276.048 -6512.4167 1069.6480 -1.3723415
0.0000000 -2021700.0
1 0.0000000 0.00
474.00000 -304853.37 1621.9579 -246.65982 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
H2O_Ice(s)
4 1 1.0 2.0 0.0
273.00000 -303622.94 198.23209 -36.246000 0.0000000
0.0000000 0.0000000
1 0.0000000 0.00
###########################################################################
Date : Sep.2013
Originator : KH
Serial No : WATER.DAT
Quality Status : A
ChemSage Vers. : 4.0
Temperatures : From approx. 270 K to about 3000 K
Compositions : Aqueous phase with gas and solid
Applications : Gas-Liquid-Solid equilibria of water with non-ideal
gas and liquid with phase internal equilibria
This file serves as an example of input for non-ideal
gas using Tsonopoulos data (Tc,Pc,Vc, acentric factor and
dipol moment) as well as Ideal Water solution with
neutral and ionic species.
Sources of data:
Unary : 2013FACT
Binary : n.a.
References : 2013FACT FACT Pure Substance database Edition 2012
###########################################################################
14.8 SUBL-EX.DAT
Directory
Contents of data-file subl-ex.dat System components: 1: Co 2: Cr 3: Fe Phases: 1: BCC#1 2: BCC#2 3: SIGMA:30#1 4: SIGMA:30#2 Phase constituents of mixture phases: Phase: BCC#1 Model: RKMP Status: ENTERED 1: Co 2: Cr 3: Fe Phase: BCC#2 Model: RKMP Status: ENTERED 1: Co 2: Cr 3: Fe Phase: SIGMA:30#1 Model: SUBL Status: ENTERED 1: Co:Cr:Co 2: Co:Cr:Cr 3: Co:Cr:Fe 4: Fe:Cr:Co 5: Fe:Cr:Cr 6: Fe:Cr:Fe Phase: SIGMA:30#2 Model: SUBL Status: ENTERED 1: Co:Cr:Co 2: Co:Cr:Cr 3: Co:Cr:Fe 4: Fe:Cr:Co 5: Fe:Cr:Cr 6: Fe:Cr:Fe
Data-file
Co-Cr-Fe See commentary below! GTT July 1999
3 5 0 3 3 6 6 0
Co Cr Fe
58.9332 51.9960 55.8470
4 1 2 4 5
4 1 2 4 5
BCC
RKMP
Co
1 1 1.0 0.0 0.0
2000.0000 6953.1800 0.00000000 -.63137000E-02 0.28037000E-05
Cr
1 1 0.0 1.0 0.0
2000.0000 0.00000000 0.00000000 0.00000000 0.00000000
Fe
1 1 0.0 0.0 1.0
2000.0000 0.00000000 0.00000000 0.00000000 0.00000000
2
1 2 3
30965.780 -27.614000 0.00000000 0.00000000
11865.820 8.2801000 0.00000000 0.00000000
20145.960 0.00000000 0.00000000 0.00000000
2
1 3 2
-38417.500 0.00000000 0.46870000E-01 -.19131800E-04
-1472.7700 0.00000000 -.31652000E-02 0.25690000E-05
2
2 3 2
25104.000 -11.427300 0.00000000 0.00000000
0.00000000 -1.2041600 0.00000000 0.00000000
3
1 2 3 3
-16736.000 12.552000 0.00000000 0.00000000
-16736.000 12.552000 0.00000000 0.00000000
-16736.000 12.552000 0.00000000 0.00000000
0
BCC
RKMP
Co
1 1 1.0 0.0 0.0
2000.0000 6953.1800 0.00000000 -.63137000E-02 0.28037000E-05
Cr
1 1 0.0 1.0 0.0
2000.0000 0.00000000 0.00000000 0.00000000 0.00000000
Fe
1 1 0.0 0.0 1.0
2000.0000 0.00000000 0.00000000 0.00000000 0.00000000
2
1 2 3
30965.780 -27.614000 0.00000000 0.00000000
11865.820 8.2801000 0.00000000 0.00000000
20145.960 0.00000000 0.00000000 0.00000000
2
1 3 2
-38417.500 0.00000000 0.46870000E-01 -.19131800E-04
-1472.7700 0.00000000 -.31652000E-02 0.25690000E-05
2
2 3 2
25104.000 -11.427300 0.00000000 0.00000000
0.00000000 -1.2041600 0.00000000 0.00000000
3
1 2 3 3
-16736.000 12.552000 0.00000000 0.00000000
-16736.000 12.552000 0.00000000 0.00000000
-16736.000 12.552000 0.00000000 0.00000000
0
SIGMA:30
SUBL
Co:Cr:Co
1 1 .8667 .1333 0.0
2000.0000 11293.367 -5.0583333 0.00000000 0.00000000
Co:Cr:Cr
1 1 .3333 .6667 0.0
2000.0000 -207.30000 -6.8573333 0.00000000 0.00000000
Co:Cr:Fe
1 1 .3333 .1333 .5334
2000.0000 2998.5333 0.00000000 0.00000000 0.00000000
Fe:Cr:Co
1 1 .5334 .1333 .3333
2000.0000 -5112.1667 0.00000000 0.00000000 0.00000000
Fe:Cr:Cr
1 1 0.0 .6667 .3333
2000.0000 4841.3333 -5.3766667 0.00000000 0.00000000
Fe:Cr:Fe
1 1 0.0 .1333 .8667
2000.0000 4040.6667 -1.9005667 0.00000000 0.00000000
3
0.333333 0.133333 0.533334
2 1 3
Co Fe
Cr
Co Cr Fe
1 1 1 2 2 2
1 1 1 1 1 1
1 2 3 1 2 3
0
SIGMA:30
SUBL
Co:Cr:Co
1 1 .8667 .1333 0.0
2000.0000 11293.367 -5.0583333 0.00000000 0.00000000
Co:Cr:Cr
1 1 .3333 .6667 0.0
2000.0000 -207.30000 -6.8573333 0.00000000 0.00000000
Co:Cr:Fe
1 1 .3333 .1333 .5334
2000.0000 2998.5333 0.00000000 0.00000000 0.00000000
Fe:Cr:Co
1 1 .5334 .1333 .3333
2000.0000 -5112.1667 0.00000000 0.00000000 0.00000000
Fe:Cr:Cr
1 1 0.0 .6667 .3333
2000.0000 4841.3333 -5.3766667 0.00000000 0.00000000
Fe:Cr:Fe
1 1 0.0 .1333 .8667
2000.0000 4040.6667 -1.9005667 0.00000000 0.00000000
3
0.333333 0.133333 0.533334
2 1 3
Co Fe
Cr
Co Cr Fe
1 1 1 2 2 2
1 1 1 1 1 1
1 2 3 1 2 3
0
###########################################################################
Date : July 1999
Originator : KH
Serial No : SUBL-EX.DAT
Quality Status : B
ChemSage Vers. : 4.0
Temperatures : 900 - 1200 K
Compositions : Iron rich or chromium rich
Applications : SIGMA-BCC equilibria
This file serves as an example of input for
substitutional solutions (BCC) described with the
RKMP model and a multi-sublattice phase (SIGMA) with
three sublattices and 30 atoms to the unit cell.
It also serves as a data-file for examples on how to
get compositional data for sublattice constituents
or overall mole fractions in case of a multi-
sublattice phase when using ChemApp.
Note the scaling of the SIGMA phase (division of stoichio-
metries and Gibbs energy coefficients by 30) to obtain
equal size of all phases.
NOTE the change of the compound names in the SIGMA phase
according to the SGTE notation for sublattice phases.
Old name New name
(Co26Cr4):30 Co:Cr:Co
(Co10Cr20):30 Co:Cr:Cr
(Co10Cr4Fe16):30 Co:Cr:Fe
(Co16Cr4Fe10):30 Fe:Cr:Co
(Cr20Fe10):30 Fe:Cr:Cr
(Cr4Fe26):30 Fe:Cr:Fe
Sources of data:
Unary : 81Zha
Binary : 81Zha
References : 81Zha Zhangpeng, J.; Jansson, B.: TRITA-MAC-0189, RIT,
Stockholm, May 1981
###########################################################################
15 Revision history
This chapter lists the main changes and additions that have been made to the ChemApp Programmer's Manual since Edition 3.1. Note that not all editions have been made available publically or been published in printed form.
Edition 3.14 (release date: November 4, 2013)
- Documentation for subroutine TQCONF has been added. To provide a useful code example, a new sample thermochemical data-file is included with ChemApp (water.dat, see WATER.DAT (System O-H-EA)).
- A new error message (Error code 312) has been added ("<NAME> is a system component, but without charge").
- Updated Table List of available solution models in ChemApp, added literature references to Table Identifiers of phase model names as returned by TQMODL.
Edition 3.13 (release date: January 20, 2010)
- A new error message (Error code 110) has been added: when thermochemical data-files with syntactic errors are read, ChemApp now reports the phase or phase constituent in which the error is likely to be found.
Edition 3.12 (release date: January 31, 2008)
- Updated manual with respect to ChemApp 'light' in various places.
- Updated references in Table List of available solution models in ChemApp.
Edition 3.11 (release date: January 21, 2008)
- Added Table Available thermodynamic data options.
- Added literature reference of new ChemApp article [Pet07].
- Initial documentation of the subroutines for data manipulation (TQGDAT, TQLPAR, TQGPAR, TQCDAT, TQWASC) have been included in the public version.
Edition 3.10 (release date: June 2, 2006)
- Added documentation for new subroutine TQCNSC.
- Added options 'pH' and 'Eh/V' to Table State variables for which results can be obtained with the subroutine TQGETR.
Edition 3.9 (release date: March 22, 2006)
- Added documentation for two new subroutines: TQCHAR and TQBOND.
- Added options 'AT', 'XT', and 'XPT' to Table Possible state variables for setting conditions in the subroutines TQSETC and TQSTEC.
- Added note to TQCEN about computation time with SUBG, SUBS, and QUAS models.
- Clarified documentation for TQCLIM with respect to units used.
- TQSTTP: stream names may now be 150 characters long.
Edition 3.8 (release date: February 15, 2005)
- The table describing the options to TQGDPC and TQSTXP has been split into two (Tables Thermodynamic quantities that can be calculated using TQGDPC and Thermodynamic quantities that can be calculated using TQSTXP), to help clarify which units are used for the returned values.
- Added a note to the TQCLIM documentation that the values are entered using bar, K, and dm3 as units, not in the "current system units".
Edition 3.6 (release date: May 1, 2003)
- Descriptions for the following new subroutines have been added: TQCEN, TQCENL, TQGTHI, and TQGTED.
- Table ` State variables for which results can be obtained with the subroutine TQGETR`_: added new option 'AP'.
- In Table List of available solution models in ChemApp and Identifiers of phase model names as returned by TQMODL, the solution models HTSZ, HTWZ, HTDZ, and PIHZ have been added.
- Added a note to Installation and use about all Windows versions of ChemApp requiring a hardware key (dongle) as of V5.0.0.
- Chapter ChemApp best practices has been added.
Edition 3.5 (release date: July 1, 2001)
- Descriptions for five new subroutines have been added that were introduced with ChemApp V3.3.1 and relate to sublattice phases: TQINLC, TQGNLC, TQNOSL, TQNOLC, TQGTLC.
- The data-file SUBL-EX.DAT (see SUBL-EX.DAT (System Co-Cr-Fe)) is now part of the ChemApp distribution. Its main purpose is to serve as an example data-file for the demonstration of the various sublattice model subroutines.
- Descriptions for the subroutines TQOPNA and TQOPNB have been added, those for TQOPEN, TQCLOS, TQRFIL, and TQRBIN have been updated accordingly. The Chapters ASCII data-files and Binary data-files (DEPRECATED) have been added to illustrate the use of the file I/O subroutines.
- The use of TQOPNA, TQOPNB, TQOPEN, and TQCLOS intead of OPEN and CLOSE is now also recommended and supported for FORTRAN programs. While this is not necessary for FORTRAN programs which are linked to ChemApp in form of a static library, it is required in many cases where ChemApp is used in form of a DLL. The documentation of the subroutines involved has been updated accordingly. Since these subroutines are now part of the ChemApp core code, the associated Error code 901 and Error code 902 have been changed to Error code 107 and Error code 108.
- A short text on user-defined models (Chapter User-defined solution models) has been added.
- In Table List of available solution models in ChemApp and Identifiers of phase model names as returned by TQMODL, the solution models SITZ, SUBE, and SUBG have been added. The models RKXP, MARP, KKOP, and KKXP are no longer available as of ChemApp V4.0.0, their entries in the tables have been deleted.
- Descriptions for the new subroutines have been added that are related to transparent file handling: TQGTID, TQGTNM, TQGTPI, TQOPNT, TQRCST, and TQGTRH. Chapter Transparent data-files has been added.
- A description for the new subroutine TQUSED has been added.
Edition 3.4 (release date: March 22, 1999)
- Table State variables for which results can be obtained with the subroutine TQGETR: added missing option 'VM' for variable Volume/amount unit. All other variables of the same group ('CPM', 'HM', 'SM', and 'GM') should be labelled per amount unit instead of per mass unit too.
Edition 3.3 (release date: February 3, 1999)
A note has been added to the description of TQCSC that it is not possible to change the system components if the system (i.e. the data-file loaded) contains one or more phases modeled using the SUBO (Two-sublattice order/disorder) formalism. A note has been added to the description of TQCSPC to explain some restrictions that apply to changing the status of phase constituents. Table Possible state variables for setting conditions in the subroutines TQSETC and TQSTEC: option 'A': For a precipitation target, VAL must be < 0, not <= 0.
Edition 3.2 (release date: January 27, 1999)
First printed and distributed edition for ChemApp versions 2.0.2 and later.
16 Alphabetical list of ChemApp subroutines
17 Bibliography
| [Bel92] | (1, 2) A.B. Belonoshko and S.K. Saxena. A unified equation of state for fluids of C-H-O-S-Ar composition and their mixtures up to very high temperatures and pressures. Geochimica et Cosmochimica Acta, 56:3611,1992. |
| [Bla87] | (1, 2, 3) M.Blander and A.Pelton. Thermodynamic analysis of binary liquid silicates and prediction of ternary solution properties by modified quasichemical equations. Geochimica et Cosmochimica Acta, 51:85,1987. |
| [Cha01] | (1, 2) P.Chartrand and A.Pelton. The Modified Quasi-chemical Model: Part III. Two Sublattices. Metall. Mater. Trans. A, 32:1397, 2001. |
| [Dav62] | (1, 2) C.W. Davies. Ion Association, Butterworths, London, 1962. |
| [Des91] | (1, 2) Y.Dessureault and A.Pelton. J. Chim. Phys., 88:1811, 1991. |
| [Eri90] | G.Eriksson and K.Hack. ChemSage - A Computer Program for the Calculation of Complex Chemical Equilibria. Metallurgical Transactions B, 21B:1013, 1990. |
| [Eri94] | G.Eriksson, H.Sippola, and B.Sundman. A Proposal for a General Thermodynamic Software Interface. In A.Jokilaakso, editor, Proceedings of the Colloquium on Process Simulation, pages 67-103, Espoo, Finland, 1994. Helsinki University of Technology. Report TKK-V-B99. |
| [Eri95] | G.Eriksson, P.J.Spencer, and H.Sippola. A General Thermodynamic Software Interface. In A.Jokilaakso, editor, Proceedings of the 2nd Colloquium on Process Simulation, page 113, Espoo, Finland, 1995. Helsinki University of Technology. Report TKK-V-B104. |
| [Eri97] | G.Eriksson, K.Hack, and S.Petersen. ChemApp - A programmable thermodynamic calculation interface. In J.Hirsch, editor, Werkstoffwoche '96, Symposium 8: Simulation, Modellierung, Informationssysteme, (1997) p.47, ISBN 3-88355-236-4, published by: DGM Informationsgesellschaft mbH, Hamburger Allee 26, D-60486 Frankfurt, Germany. |
| [Gay92] | (1, 2) H.Gaye and J.Lehmann and T.Matsumiya and W.Yamada. Modèle Thermodynamique des laitiers: Application à des systèmes contenant S, F, P2O5 et des oxydes de Chrome. IRSID, Maizières-lès-Metz, France. Fiche de recherche EGN 194, 1992. |
| [Gre97] | (1, 2) I.Grenthe, A.V.Plyasunov and K.Spahiu. In I.Grenthe and I.Puigdomenech, eds., Modelling in Aquatic Chemistry, OECD NEA, Paris, 1997, p. 325. |
| [Hel81] | (1, 2) H.C. Helgeson, D.H. Kirkham, and G. C. Flowers. Theoretical prediction of the thermodynamic behavior of aqueous electrolytes at hight temperatures and pressures: IV. Calculation of activity coefficients, osmotic coefficients, and apparent molal and standard and relative partial molal properties to 600 degrees C and 5kb. American Journal of Science, 281:1249,1981. |
| [Hoc84] | (1, 2) M.Hoch and I.Arpshofen. Über ein Komplexmodell zur Berechnung der thermodynamischen Zustandsfunktion flüssiger Legierungen. Z. Metallkde., 75(1):23, 1984. |
| [Hac95] | K.Hack. ChemSage-TEACH - A Selftuition Package for Thermochemical Calculations. GTT-Technologies, 1995. |
| [Hac96] | K.Hack, editor. The SGTE Casebook -- Thermodynamics at Work. Number 621 in Materials Modelling Series. Institute of Materials, London, 1996. |
| [Hil85] | (1, 2) M.Hillert and B.Jansson. Thermodynamic model for very nonstoichiometric ionic phases - application to CeO2-x. TRITA-MAC Report 0272, Materials Research Center, The Royal Institute of Technology, Stockholm, Sweden, 1985. |
| [Har84] | (1, 2, 3, 4, 5, 6) C.Harvie, N.Møller, and J.Weare. The prediction of mineral solubilities in natural waters: The Na-K-Mg-Ca-H-Cl-SO4-OH-HCO3-CO3-CO2-H2O system to high ionic strengths at 25 degrees C. Geochimica et Cosmochimica Acta, 48:723, 1984. |
| [Hoc84b] | (1, 2) M.Hoch. Application of the complex model to systems with an asymmetric miscibility gap and with two miscibility gaps. Z. Metallkde., 75(4):299, 1984. |
| [Hoc87] | (1, 2) M.Hoch. Application of the Hoch-Arpshofen model to ternary, quaternary, and larger systems. CALPHAD, 11(2):219, 1987. |
| [Lac91] | (1, 2) J.Lacaze and B.Sundman. An assessment of the Fe-C-Si system. Metallurgical Transactions A, 22A:2211, 1991. |
| [Oat95] | (1, 2) W.A.Oates, G.Eriksson, and H.Wenzl. Thermodynamic modelling of solid gallium arsenide. J. Alloys Compounds, 220:48, 1995. |
| [Oat] | W.A.Oates. Private communication. |
| [Pel86b] | (1, 2) A.Pelton and C.Bale. A modified interaction parameter formalism for non-dilute solutions. Metallurgical Transactions A, 17A:1211, 1986. |
| [Pel86] | (1, 2, 3) A.Pelton and M.Blander. Thermodynamic analysis of ordered liquid solutions by a modified quasichemical approach - application to silicate slags. Metallurgical Transactions B, 17B:805, 1986. |
| [Pel00] | A.Pelton, S.A.Degterov, G.Eriksson, C.Robelin, and Y. Dessureault. The Modified Quasichemical Model I - Binary Solutions. Metall. Mater. Trans. B, 31B:651, 2000 |
| [Pel] | (1, 2) A.Pelton. Private communication. |
| [Pel01] | (1, 2) A.Pelton and P.Chartrand. The Modified Quasi-chemical Model: Part IV. Two-Sublattice Quadruplet Approximation. Metall. Mater. Trans. A, 32:1409, 2001 |
| [Pel88] | (1, 2, 3, 4) A.Pelton. A database and sublattice model for molten salt solutions. CALPHAD, 12:127, 1988. |
| [Pel93] | (1, 2, 3) A.Pelton, G.Eriksson, and A.Romero-Serrano. Calculation of sulfide capacities of multicomponent slags. Metallurgical Transactions B, 24B:817, 1993. |
| [Pet07] | (1, 2) S.Petersen and K.Hack. The thermochemistry library ChemApp and its applications. Int. J. Mat. Res., 98(10):935, 2007. |
| [Pit73] | K.S.Pitzer. Thermodynamics of Electrolytes. I. Theoretical Basis and General Equations. J. Phys. Chem., 77(2):268, 1973. |
| [Red48] | (1, 2) O.Redlich and A.T.Kister. Algebraic representation of thermodynamic properties and the classification of solutions. Ind. Eng. Chem., 40(2):345, 1948. |
| [Sun81] | (1, 2) Sundman and J.Ågren. A regular solution model for phases with several components and sublattices, suitable for computer applications. J. Phys. Chem. Solids, 42:297, 1981. |
| [Sun91] | (1, 2) B.Sundman. CALPHAD, 15:109, 1991. |
| [Tan88] | (1, 2, 3, 4, 5, 6, 7, 8) J.C. Tanger and H.C. Helgeson. Calculation of the thermodynamic and transport properties of aqueous species at high pressures and temperatures: revised equations of state for the standard partial molal properties of ions and electrolytes. American Journal of Science, 288:19, 1988. |
| [Tso74] | (1, 2) C.Tsonopoulos. An empirical correlation of second virial coefficients. AIChE Journal, 20(2):263, 1974. |
| [Zha81] | (1, 2) J.Zhangpeng and B.Sundman. A study of the sigma phase in the Co-Cr-Fe system. TRITA-MAC Report 0189, Materials Research Center, The Royal Institute of Technology, Stockholm, Sweden, 1981. |